
1. Introduction
Impurity is considered as any component of a drug substance that is not a chemical entity defined as the drug substance and in addition, for a drug product, any component that is not a formulation ingredient [1]. Impurity profiling is the basis for determining, assuring the quality, safety, and efficacy of drug substances and drug products.
Defining the impurity profile of the drug substance (Active Pharmaceutical Ingredient) is the basis for impurity profiling of drug products, however, it also considers excipients and the formulation process. This paper will discuss the guidelines to be considered in categorizing the impurities, identifying the possible source, evaluating, analytical methods which can be used, qualifying impurity levels, and proposing limits.
Impurities in Drug substances and Drug products can be classified as follows as per the current guidances of ICH, FDA, and EU requirements.
a) Organic impurities
I. Process
II. Degradation
III. Chiral impurities
b) Genotoxic impurities
c) Inorganic impurities
d) Residual solvents
e) Polymorphic impurities
The evolution of impurities profiles testing in drug substances and drug products over the period can be largely categorized as below (Figure 1).
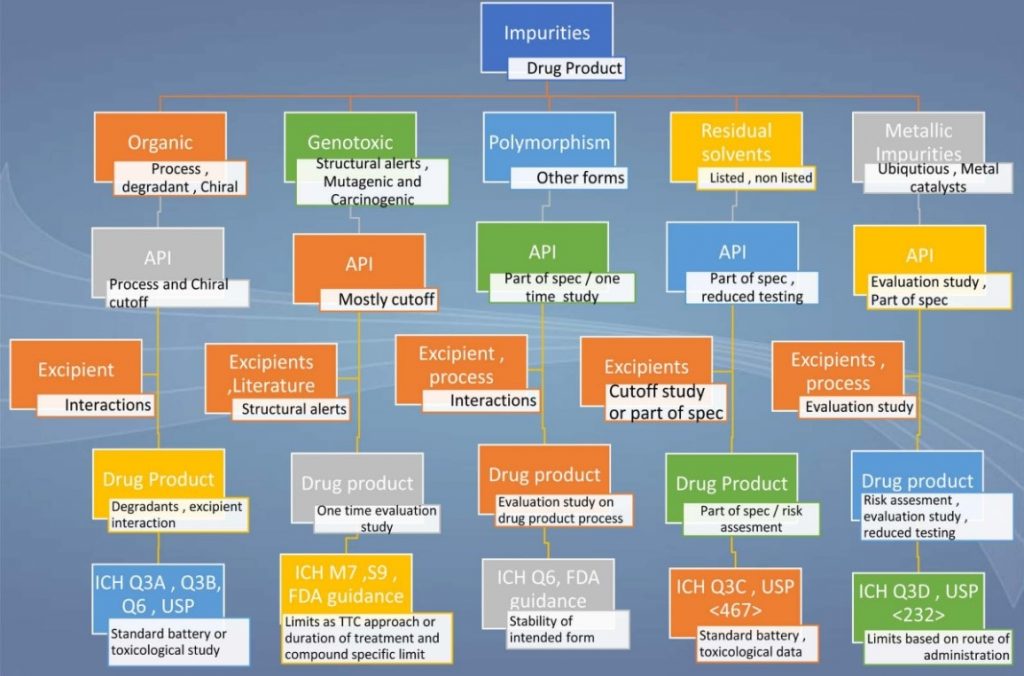
A snapshot of the impurity profiling of drug substances and drug products is presented in the below illustration (Figure 2).
2. Organic Impurities
An overview of organic impurities profiling is detailed in the following illustration Figure 3.
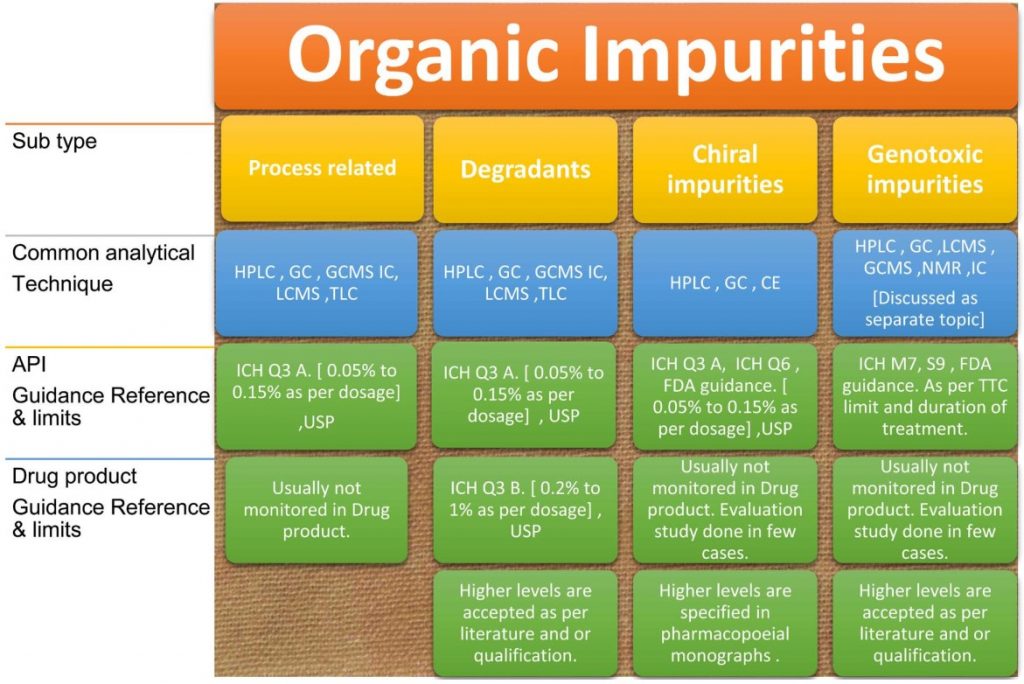
Organic impurities are broadly classified into the following categories:
2.1. Process related impurities
Starting materials, by-products, intermediates, reagents, ligands, and catalysts impurities that are not increased in stability studies and not formed during forced degradation studies are considered process-related impurities.
Process related impurities are formed during the synthesis, purification, and storage of the drug substances. Most of these impurities will be expected during the synthesis based on knowledge of the synthesis process, the type of chemical/reagents used, also these should be confirmed with stability studies [4] and degradation studies. The impurities selection should be cross verified or challenged by stress studies or force degradation studies [12].
Impurities in final product specifications may be proposed based on the impurities detected in commercial processes for setting better process control. Impurities that are above the qualification limits should be considered in specification with reporting and qualification thresholds based on their safety levels.
Whereas in drug products demonstrating specificity (usual separation in chromatographic methods) for process-related impurities with degradation impurities is the requirement for the stability-indicating method but there is no requirement to report process-related impurities (as they are controlled in applicable raw materials) unless it is mentioned in the respective pharmacopeial monograph.
2.1.1. Analytical techniques [31]
Advanced techniques for quantification of these impurities are High-Performance Liquid Chromatography (HPLC), Ultra Performance Liquid Chromatography (UPLC)/ Rapid resolution liquid chromatography (PPLC), Gas Chromatography (GC), Liquid chromatography-Mass spectrometry (LC-MS), Gas Chromatography-Mass Spectrometry (GC-MS).
The limit is governed by ICH Q3A “Impurities in New Drug Substances”.
2.1.2. Drug Substances Specification [2]
Maximum daily dose: ≤ 2 g/day: 0.15% or 1.0 mg per day intake (whichever is lower).
Maximum daily dose: > 2 g/day: 0.05%
2.1.3. Drug product specification
Not considered for reporting unless it is mentioned in a specific pharmacopeia monograph.
2.2. Degradation Impurities/Products
Forced degradation studies should be performed to identify potential degradation products, characterization of a degradation product, stability of the active molecule, understanding the degradation pathways of the drug, rate of degradation, develop stability-indicating analytical method, and define shelf life.
Impurities that are increased during stability studies (Q1A) and/or formed during forced degradation studies are classified as degradation impurities/products.
The selection of conditions for degradation studies and levels of target degradation is practiced as per common industry practices and available peer-reviewed publications. ICH guidance Q1B “Photostability testing of new drug substances and products” serves as a guidance document on photostability [13].
2.2.1. Analytical techniques
HPLC-PDA/UV detector, GC, LC-MS, and GC-MS techniques shall be employed in the identification of impurities with a validated analytical procedure [5].
As per the current requirements and recent deficiencies, forced degradation studies, peak purity studies, and mass balance for individual impurity quantifying methods should be performed. Mass balance is “the process of adding together the assay value and levels of degradation products to see how closely these add up to 100% of the initial value, with due consideration of the margin of analytical precision” [15].
Impurities found above identification threshold should be considered in evaluating impurity profile for drug substance/drug product.
2.2.2. Drug Substance Specification [2]
Maximum daily dose: ≤ 2 g/day: 0.15% or 1.0 mg per day (whichever is lower)
Maximum daily dose: >2 g/day: 0.05%.
Performing individual excipient and drug compatibility studies is one of the recommended approaches to assess degradation pathways in Drug products. These studies should be designed as per the available literature and also known behavior for some excipients like povidone, silicon dioxide, Lactose, Polysorbate, etc [14] [15].
Oxidative and photolytic conditions require special considerations to understand the mechanism of photodegradation and the potential for phototoxicity [18]. Photostability testing should be performed as per ICH Q1B as an integral part of stress testing [13].
2.2.3. Drug Product Specification [3]
Maximum daily dose: <10 mg: 1.0% or 50 µg of TDI (Total daily intake) (whichever is lower).
Maximum daily dose: 10 mg – 100 mg: 0.5% or 200 µg of TDI (whichever is lower).
Maximum daily dose: >100 mg – 2 g: 0.2% or 3 mg of TDI (whichever is lower).
Maximum daily dose: >2 g: 0.15%.
High levels of impurities are acceptable with justification i.e. with literature. and qualification.
2.3. Chiral Impurities
Enantiomers, non-superimposable mirror image stereoisomers with identical Physico-chemical properties except that they rotate the plane of polarized light in opposite directions and equal amounts [16].
If one of the enantiomers is considered as the active ingredient, then the other may/shall be considered as impurity/undesired.
In earlier days, there was not much focus on individual enantiomers and were not well studied or characterized due to the limitation of analytical techniques. Current advanced techniques allow to define, study completely, separate, and quantify stereoisomeric impurities [17].
If applicable, stereoisomeric purity of starting material should be studied with a well resolved and validated stereoselective test procedure and possible formation of stereogenic center in individual synthetic process details and controls should be established [19].
For a drug substance stability indicating method, it is required to perform forced degradation studies for chiral impurity methods and confirm its formation. If the chiral impurity increases instability studies, then it should be monitored in drug products.
2.3.1. Analytical techniques
Advanced analytical techniques used in chiral separation are Gas Chromatography (GC), High Performance Liquid Chromatography (HPLC), UHPLC, Capillary Electrophoresis (CE), and liquid phase Nuclear Magnetic Resonance Spectroscopy (NMR). The recent trend of emergence of Polysaccharide derived chiral stationary phases (CSPs) have been recognized as the most powerful materials for the chromatographic separation of enantiomers in analytical and preparative applications due to their broad application field and their remarkable loading capacity, a wide variety of solvents both polar and non-polar can be used to achieve desirable separations.
2.3.2. Drug substances specification
Maximum daily dose: ≤ 2 g/day: 0.15% or 1.0 mg per day (whichever is lower).
Maximum daily dose: > 2 g/day: 0.05%.
In most cases, chiral impurities are controlled in drug substances but in some cases, control of chiral impurities and confirmation of an unacceptable change in stereochemical purity or ratio of the active substance that occurs in drug product shelf life should be established with a validated analytical method [20].
2.3.3. Drug product specfication
In general, not included in the specification, but in some cases, evaluation may be required.
3. Inorganic impurities
Inorganic impurities can result from the manufacturing process. They are normally known and identified and include reagents, ligands, and catalysts, heavy metals or other residual materials, Inorganic salts, Other materials (e.g. filter aids, charcoal) [1]. Heavy metals test by color comparison been a part of USP since 1904, due to numerous events on the health of the patient population the controls on metallic impurities are effective and in full control from Jan. 1st, 2018 across all the major regulatory authorities.
3.1. Elemental impurities
Based on decades of studies, knowledge of toxicological effects and to improve the safety and efficacy of drugs, current requirements changed and replaced nonspecific heavy metal tests with specific quantifying techniques [21] and specifications [22] were included. The classical color comparison test in Pharmacopeia is deficient in detecting a few metallic impurities. The standard solutions of all the metals are also of different solubilities due to the variable levels of reactivities of metals with sulfide ions, which also creates a problem in visual comparison with lead standards as shown in Figure 5 [23].

Process catalysts and environmental contaminants may be present in drug substances, excipients, or drug products. These impurities may occur naturally, be added intentionally, or be introduced inadvertently. As elemental impurities do not provide any therapeutic benefit to the patient, besides continuous exposure might be toxic (as the toxicity is related to its extent of exposure), their toxic levels in the drug product should be controlled within acceptable limits [24].
3.1.1. Risk assessment
Elemental impurities are classified into 4 categories [1, 2A, 2B, and 3] based on the route of administration, limits, and requirements for control varies. Risk assessment should be done for elements: Cd, Pb, As, Hg, Co, V, Ni, Tl, Au, Pf, Ir, Os, Ph, Ru, Se, Ag, Pt, Li, Sb, Ba, Mo, Cu, Sn, Cr as per ICH Q3D [6] and USP <232> based on potential source, Evaluation of toxicity data concerning the route of administration, Establishment of a Permitted Daily Exposure (PDE) and Justification for higher levels than established PDE.
In drug product risk evaluation, potential sources of elemental impurities that should be considered are elements intentionally added such as process catalysts, elements potentially present in the materials used to prepare the drug product, and elements potentially introduced from manufacturing equipment or container closure systems [24] [6].
Based on risk assessment, it should be concluded to include testing of elemental impurities in routine testing for components of drug products or drug products i.e. if elemental impurities are controlled in raw material (active and inactive) as part of routine testing, in drug product routine testing may not be required. ICH Q3D defines different options (Option 1, Option 2a, Option 2b, and Option 3) for risk assessment and control strategy (If batch analysis results are consistently less than 30% of the PDE then additional controls are not required).
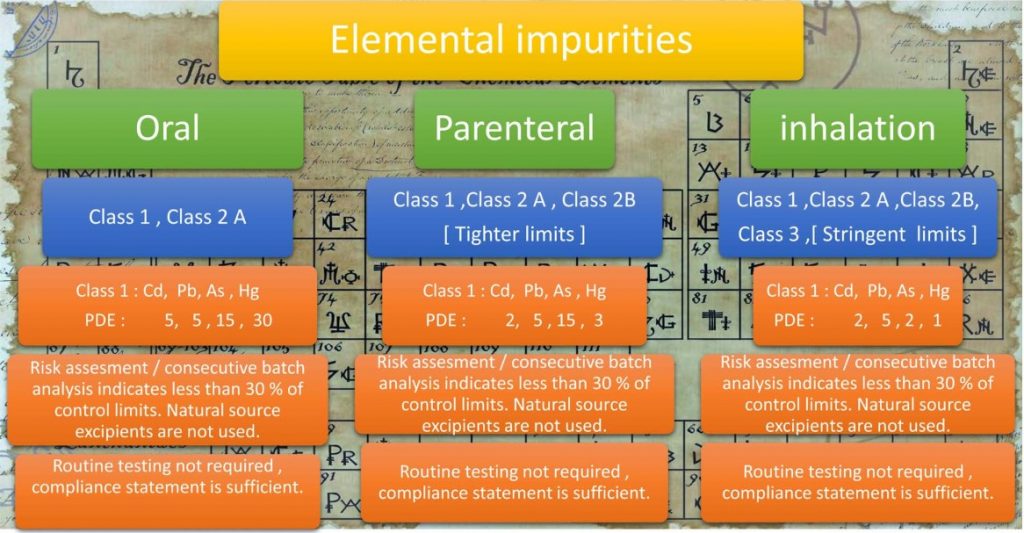
An overview of elemental impurities as per ICH Q3D to consider for risk assessment is presented below in Figure 6.
3.1.2. Specifications
ICH Q3D classifies metallic impurities into Class 1, 2A, 2B, and 3 with decreasing levels of toxicity. The specifications and class of impurities to be controlled vary on the route of administration.
3.1.3. Analysis Techniques
In general, typical analytical techniques followed for elemental impurities analysis are ICP, and ICP-MS. These can be analyzed with the method listed in USP <233>, if alternate methods are used those are to be validated as per ICH Q2, USP <1225>. There are few alternate analytical methods available and reported in research articles [23].
4. Residual Solvents
Residual solvents are organic liquids used as vehicles for the preparation of solutions or suspensions in the synthesis of a drug substance or excipients or the preparation of a drug product.
The residual solvents used in each stage of the manufacturing process of Drug substance or excipient can either be analyzed and exhibited that they meet the ICH Q3C [7], USP <467> requirements [8] or they can be analyzed in the final stage, in either case they should be analyzed with validated method or method specified in USP <467> [8]. The residual solvents with known limits and toxicity details concerning their class were listed in ICH Q3C, their specifications in the final product should be maintained to the possible lowest extent (The acceptance limits should be based on ICH Q3C, USP <467>). If non-listed solvents are used they should be controlled as per respective solvent toxicological limit.
In recent revision of ICH Q3C, Triethylamine is added as Class-3 solvent with a PDE limit (62.5 mg/Day), and the limit for Methyl isobutyl ketone is revised (Class-2) PDE limit (45 mg/day) as per the new toxicological data.
4.1. Analytical techniques
Usually, GC and GC-HS methods are employed and, in some cases, HPLC, IC, and other techniques are employed. If residual solvent method is specified in a monograph, the same can be followed or the method specified in USP <467> can be used. USP <467> does not specify a method for class-3 solvents but FDA cites to control and report class 3 solvents also with a validated method.
4.2. Drug Substance Specifications
As per ICH Q3C classification.
4.3. Drug Product Specification
As the residual solvents limits were set as per the guidelines and quantified in individual excipient and drug substances, testing for solvents in drug products is not required. Compliance may be demonstrated through comprehensive risk assessment based on quantified results of residual solvents present in individual drug substances and excipients [25].
If residual solvents are used in the manufacturing process of drug products, they should be quantified with validated analytical methods with ICH Q3C limits.
If a non-listed solvent is used in the process, then the justification for limits proposed, control strategy, and toxicity evaluation should be presented/executed.
5. Polymorphic Forms/Impurities
Polymorphism evaluation shall be used for estimation of amorphous and crystalline of qualitative and quantitative as per USP <941>, ICH Q6A, and FDA guidance “ANDAs: Pharmaceutical Solid Polymorphism” [26] [27].
As the physical properties of raw material may change with polymorphic form, affect the manufacturing process of the drug substance and the drug product, as well as drug product stability, dissolution, and bioavailability, the type and extent of characterization and release testing performed on the co-crystal should be sufficient to ensure the identity, strength, quality, and purity of the API(s) and drug product stability [54] [55] [56]. Any polymorphic form other than desired polymorphic form will be considered an impurity [57].
Based on the process and evaluation done by drug substance manufacturers, a polymorphic form will be confirmed.
In drug products, drug substance polymorphic form should not change due to the drug product manufacturing process or due to excipients or storage over some time. Drug product initial samples and stability samples should be evaluated to prove the polymorphic form stability throughout the shelf life [27]. Information on different polymorphs of drug substances is generally referred to in respective patents.
5.1. Analytical Techniques
A common analytical technique used is polymorphism by p X-ray diffraction. The method adopted should be specific to desired crystalline form of the compound based on the drug substance manufacturing process. Method should be validated as per USP <1225> [5].
In some cases XRD pattern of different polymorphic forms might be similar, then other techniques like Thermal analysis (DSC) or Raman spectroscopy should be used.
6. Genotoxic Impurities (Mutagenic and Carcinogenic Potential)
Genotoxic impurities are DNA reactive substances that have the potential to directly cause DNA damage when present at low levels leading to mutations and therefore, potentially causing cancer. This type of mutagenic carcinogen is usually detected in a bacterial reverse mutation (mutagenicity) assay. Structure-based assessments are useful for predicting bacterial mutagenicity outcomes based on established knowledge. There are a variety of approaches to conducting this evaluation, including a review of the available literature and/or computational toxicology assessment [60].
ICH M7 definition of genotoxic impurities does not apply to drug substances and drug products intended for advanced cancer indications as defined in the scope of ICH S9 [10] [11]. Additionally, there may be some cases where a drug substance intended for other indications is itself genotoxic at therapeutic concentrations and may be expected to be associated with increased cancer risk. Exposure to a mutagenic impurity in these cases would not significantly add to the cancer risk of the drug substance. Therefore, impurities could be controlled at acceptable levels for non-mutagenic impurities [29].
Impurities below the identification limit with structural alerts (e.g., N-nitroso-, and alkyl-azoxy compounds), and potential impurities should be considered for genotoxic/mutagenic evaluation [28]. This risk-based assessment of individual synthetic processes should be performed by considering starting material/reagents used in the process and intermediate/byproducts formed during the synthesis process [29]. The suitability of the proposed control strategy can be supported with information about any mutagenic impurities formed or purged in the manufacturing steps between the proposed starting material and the drug substance, or that are controlled in the specification of the proposed starting material [28].
6.1. Analytical Techniques
LCMS, GC-MS are normally used for genotoxic impurity detection and quantification to achieve the desired limit of quantification. HPLC, UPLC, and GC methods may be used in some cases.
6.2. Threshold of Toxicological Concern (TTC)
The threshold of Toxicological Concern (TTC) concept was developed to define an acceptable intake for any unstudied chemical that poses a negligible risk of carcinogenicity or other toxic effects. The methods upon which the TTC is based are generally considered to be very conservative since they involve a simple linear extrapolation from the dose giving a 50% tumor incidence (TD50) to 1 in 106 incidence cases, using TD50 data for the most sensitive species and most sensitive site of tumor induction [10].
The concentration limits in ppm of genotoxic impurity in drug substances derived from the threshold of toxicological concern (TTC) can be calculated based on the expected daily dose to the patient using equation [10] [30].
Concentration limit (ppm) = TTC [μg/day]/dose (g/day)
An overview of the genotoxic impurity guidance is presented in Figure 7.
ICH guidance M7 classifies possible genotoxic impurities into 5 classes, which are presented in Table 1 below.
Standard risk assessments of known carcinogens assume that cancer risk increases as a function of cumulative dose. Thus, the cancer risk of a continuous low dose over a lifetime would be equivalent to the cancer risk associated with an identical cumulative exposure averaged over a shorter duration.
ICH M7 defines limits for a single impurity and a total of two or more impurities as per the duration of treatment as per below Figure 8 limits [10].
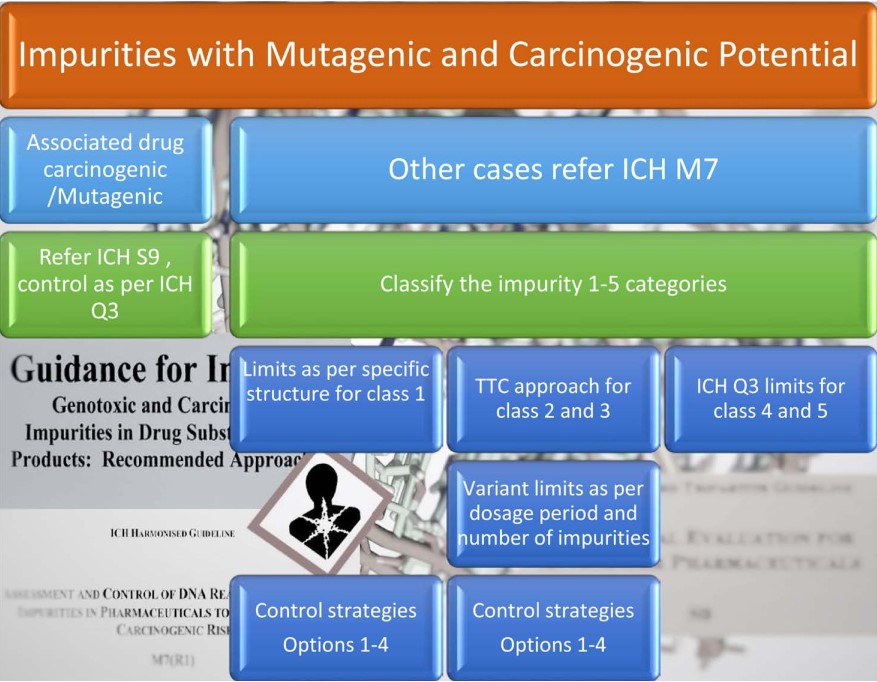
Table 1. Genotoxic impurities as per ICH M7 classified into five classes [10]

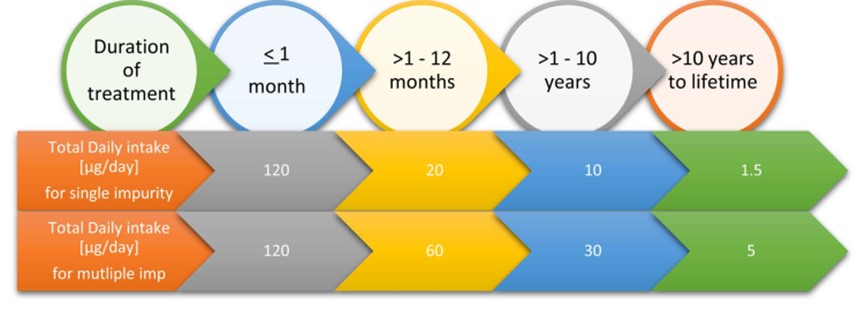
ICH M7 defines different control strategies with Options 1-5. Option 3 allows manufacturers to avoid release testing if the impurities are less than 30% of control in a few batches.
Pharmacopeias are embracing this concept slowly however regulatory authorities are watchful and issuing deficiency to control the impurities. Bromochloropropionphenone impurity in Bupropion Hydrochloride, USP specifies this impurity at NMT 0.1% but as per literature impurity should be controlled at 4 ppm, FDA has cited this as a deficiency for the filers.
6.3. Specifications
As per ICH M7 and S9 or respective impurity toxicological limits.
7. Keywords and sources
Keywords
Impurity profile, Degradation, Stereo Isomers, Genotoxic, Polymorphic, Elemental Impurities, Organic Impurities, Inorganic Impurities, Specification.
Sources
The main content of this post was quoted from the article of Pannala, R. (2018), Impurity Profiling of Solid Oral Drug Products to Sail through GDUFA-II Requirements, American Journal of Analytical Chemistry, 9, 187-209.
8. Reference
- USP <1086>, Impurities in Drug substances and drug products.
- ICH Q3A Impurities in New Drug Substances.
- ICH Q3B Impurities in New Drug Products.
- ICH Q1A Stability Testing of New Drug Substances and Products.
- ICH Q2 Validation of Analytical Procedures.
- ICH (2014) Guideline for Elemental Impurities Q3D. ICH Harmonized Guideline.
- ICH Q3C Impurities: Residual Solvents
- USP <467> Residual Solvents.
- ICH Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances
- ICH M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in pharmaceuticals to Limit Potential Carcinogenic Risk.
- ICH S9 Nonclinical Evaluation for Anticancer Pharmaceuticals.
- Sharp, T.R. (Presentation) Forced Degradation: What? Why? How? FreeThink Technologies, Inc., Groton, Connecticut.
http://www.cbinet.com/sites/default/files/files/Workshop%20B(3).pdf - ICH Q1B, Photostability Testing of New Substances and Products
- Fathima, N., Mamatha, T., Qureshi, H.K., Anitha, N. and Rao, J.V. (2011), Drug-Excipient Interaction and Its Importance in Dosage form Development. Journal of Applied Pharmaceutical Science, 1, 66-71.
- Bharate, S.S., Bharate, S.B. and Bajaj, A.N. (2010) Interactions and Incompatibilities of Pharmaceutical Excipients with Active Pharmaceutical Ingredients: A Comprehensive Review. Journal of Excipients and Food Chemicals, 1, 3-26.
- European Medicines Agency (1994) Investigation of Chiral Active Substance. European Medicines Agency Guideline.
- FDA (1992) Development of New Stereoisomeric Drugs. FDA Guidance.
- Janzen, H. (2016) Forced Degradation Studies—Comparison between ICH, EMA, FDA and WHO Guidelines and ANVISA’s Resolution RDC 53/2015. Master of Drug Regulatory Affairs, Rheinische Friedrich-Wilhelms-University of Bonn, Bonn.
- Karnes, H.T. and Sarkar, M.A. (1987) Enantiomeric Resolution of Drug Compounds by Liquid Chromatography. Pharmaceutical Research, 4, 285- 292.
https://doi.org/10.1023/A:1016437018323 - Canada Health (2000) Guidance on Stereochemical Issues in Chiral Drug Development.
- USP <233> Elemental Impurities-Procedures.
- USP <232> Elemental Impurities-Limits.
- Raghuram, P., Soma Raju, I.V. and Sriramulu, J. (2010) Heavy Metals Testing in Active Pharmaceutical Ingredients: An Alternate Approach. Pharmazie, 65, 15-18.
- CDER (2016) Elemental Impurities in Drug Products Guidance for Industry. Draft Guidance
- CDER (2009) Guidance for Industry Residual Solvents in Drug Products Marketed in the United States
- USP <941> X-Ray Powder Diffraction.
- CDER (2007) Guideline on ANDAs: Pharmaceutical Solid Polymorphism Chemistry, Manufacturing, and Controls Information.
- Benigni, R. and Bossa, C. (2006) Structural Alerts of Mutagens and Carcinogens. Current Computer-Aided Drug Design, 2, 169-176.
http://www.iss.it/binary/meca/cont/Ccadd2006%20.1161263198.pdf - ICH Q11 Guideline: Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities). Questions and Answers Version, 23 August 2017.
- EMEA (2007) Guideline on the Limits of Genotoxic Impurities.
- Pannala, R. (2018) Impurity Profiling of Solid Oral Drug Products to Sail through GDUFA-II Requirements. American Journal of Analytical Chemistry, 9, 187-209.


You must be logged in to post a comment.