
1. Introduction
Documentation is an essential part of the quality assurance system and proves that the GMP-relevant activities are carried out in a proper and timely manner. All relevant information is recorded clearly and in full so that the errors in interpretation or oral communication are minimized. Documentation reflects the planning, control, and evaluation of the quality of medicinal products. Additionally, it must be by the requirements described in Chapter 4 “Documentation” of the EU GMP guideline.
2. GMP-relevant documents and records
According to EU-GMP guidelines, GMP documentation consists of the following documents:
- Site Master File: A document describes the GMP-relevant activities of the manufacturer
- Instruction type (directions or requirements):
- Specifications: The detailed requirements that products or materials to be met during the manufacture,
- Manufacturing Formulae, Processing instruction, Packaging instruction, Testing instruction,
- Procedures (Standard Operating Procedures – SOPs),
- Protocols,
- Technical Agreements: Used in outsourced activities
- Record/Report type:
- Records: Provide evidence to demonstrate the compliance with requirements of instruction,
- Certificates of Analysis,
- Reports.
The structure of the documentation system is described in Figure 1 for example.

GMP documentation often consists of a pair of documents: one instruction document and another document that confirms that in practice everything was done by the instructions (i.e., record, reports). There are some differences between the two documents as follows (Table 1):
Table 1: GMP-relevant documents and records
| Documents | Records or Reports |
| Describe requirements | Record practical activities or their results Present the essential procedures in an easy-to-follow manner |
| Be created, reviewed, and approved in advance | Be recorded soon after the relevant activity took place |
| Handwritten entries may not be made | Handwritten entries may be made (according to Good Documentation Practice) |
| Example: Specifications, procedural instructions, contracts … | Example: Batch processing records, inspection reports … |
3. Control of documentation
Although the type and scope of the documents used in a pharmaceutical operation vary greatly, the following formal requirements will be applied when compiling the documents:
- The area of application and aim of the document: They describe where the document is used and for what purpose. The document may be misunderstood if one of these features is not stated properly,
- Written form: “Anything that has not been written down has not taken place”. This principle applies when establishing requirements in instruction documents and logging accomplished activities in records/reports. Handwritten entries should be made in a clear, legible, indelible way (pencil must not be used),
- Correctness: The document content should comply with the submission file for Marketing Authorization, Manufacturing Authorization, or other higher-level documents, as well as the current situation in the company. The correctness of critical data is checked by using the principle of dual control, i.e., checked by a second person,
- Completeness: All records must be completed with no gaps. Empty fields are marked with “Not applicable” or “N/A”, date, and signature. The total number of pages or a table of content may be used to ensure the completeness of documents,
- Accuracy: The content of the document must be accurate enough to assure the intended use. Therefore, vagaries should be avoided (see Table 2 for example).
- Currency: All documents and records should be current and up-to-date. This means that if any changes have been made, documents should be compiled on time as required. For logbooks and other records, they should be prepared at the time relevant activities occurred.
- Availability: The relevant documents must be available to the responsible person on-site. Any modifications to the document must be communicated to him/her as soon as possible.
- Approval: The authorized person must approve all documents and records. A prerequisite for approval of a document is its suitability of it (accuracy, topicality, practicality).
- Documents are comprehensible if readers can understand the contents of the document:
- An instruction for a less-skilled temporary worker will need to be written differently than an instruction for qualified specialist personnel,
- For higher-level documents, (e.g., quality management guidelines), it may be acceptable to explain the objective and leave the means of achieving it open,
- At the operational level, (e.g., manufacturing instructions) detailed specifications on how to achieve the objective are required and the control objective must be presented. These instructions must therefore be precise and unambiguous.
Table 2: Example of accuracy principle
| Incorrect | Correct |
| “long enough”, “sufficiently long” | x to y minutes/hours |
| “if possible” | Clear description, define exceptions |
| “if applicable”, “if required”, “where applicable” | Specify when the case applies |
| “should”, “should be”, “could” | “must be” |
| … | … |
4. Format and structure of documents
Documents should have a consistent format and structure. This will increase the degree of acceptance through increasing readability and making the document more recognizable:
- Document formatting: e.g., font style and size, line spacing, paragraph formatting, the layout of a document header and footer, borders, or titles …
- A logical structure allows for a quick overview, enhances the speed at which positions in the text can be located, makes the integration of changes easier, and thus reduces the time spent working with the document. See an example in Figure 2.

5. Life cycle of documents
Documents, like facilities or equipment, go through certain stages throughout their life cycle (Figure 3). The documents should be handled at each stage by established requirements to ensure that it is suitable for their intended use.
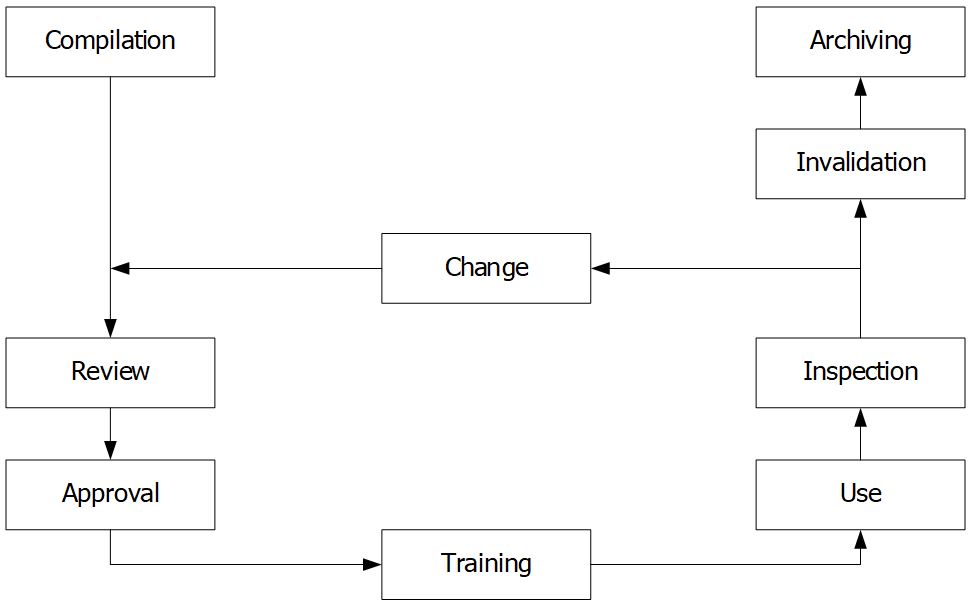
Compilation and change
Only the person who deals in practice with the provisions in the document and is knowledgeable about the procedure should create new documents or change the existing documents.
Changes can only be made once they’ve been approved and according to a specific procedure. These changes are detailed in the history of the document.
Review
The form and content of the document are reviewed by the responsible person, for example:
- Document format and structure: a responsible employee in the Quality Assurance department,
- The technical review: a person with the necessary technical expertise and experience to review the provisions set out by the compiler.
Approval
A person who is responsible for the area regulated by the document, e.g., Head of Production or Head of Quality Control.
Training
The relevant staff must know, understand and be able to apply documents. Then, he/she confirms by signature on the training form.
Use
For the term of the document’s validity, it should be kept on-site. There should be a provision stating whether personal working copies can be created and shared with third parties (if necessary). The procedure used for the distribution of new versions must be prepared.
Inspection
Determine whether the documents are being followed and whether their contents are still up-to-date and suitable.
Invalidation
It’s a good idea to establish a time restriction on how long documents may be used. As a general rule, a validity term of three years should be enough. After that time, the document can no longer be used, unless it has been formally reviewed and a new version number has been assigned. It is forbidden to use documents that are no longer valid. To avoid the unintended use of non-valid documents, they must be withdrawn.
Archiving
The storage of documents after they have been used is governed by national regulation. Data must be stored in a suitable area. Ensure that access rights to the archiving documents are restricted to authorized persons.
Handling document copies
A certain number of copies is made and distributed. Written rules must be in place, which specifies:
- who has the authority to make copies,
- how many copies may be made,
- who may receive copies (customers, authorities),
- how copies are to be marked and,
- how invalid copies are to be returned or destroyed.
6. References
- EU-GMP guideline, Part I, Chapter 4 “Documentation”
- GMP – WHO TRS 986, Annex 2: WHO-GMP for a pharmaceutical product, main principles, section 15: Documentation
- GMP – Verlag, Chapter 15 “Documentation”



You must be logged in to post a comment.