
Several important uses of Quality Risk Managementin GMP practice
Quality Risk Management is increasingly becoming an integral part of an effective pharmaceutical quality system. It can provide a proactive approach to identifying, scientifically evaluating, and controlling potential risks to quality and business. It also facilitates continual improvement and is a good tool for knowledge management. The application of QRM is very diverse and differently dependent on each organization, however, a number of risk assessments are frequently required in GMP practice.
Risk assessment for cross-contamination
The Quality Risk Management process aims to assess and control the cross-contamination risks presented by the products manufactured, hence determining the extent of technical and organizational measures required to control risks for cross-contamination. Risk assessments are usually conducted for each room or area of storage, production, and packaging based on the following plausible pathways:
- Mix-up
- Retention product contact surfaces
- Mechanical transfer
- Airborne transfer
Formalised risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal products for human use
Excipients are integrated into the drug product formulation for various reasons: to aid the drug product formulation, to serve as functional or nonfunctional ingredients, or to enable the drug to be released from the drug product in a particular, desired manner. The rationale for their use extends beyond the point of medical use to include maintenance of the safety or function of the drug product, even during storage. While good manufacturing practices (GMP) regulations clearly outline what is required for active pharmaceutical ingredients, only a basic framework exists for ascertaining appropriate GMP for excipients through risk assessment.
In recognition of the criticality of excipients in the production of drug products, the European Commission (EC) Falsified Medicines Directive (2011/62/EU) revised Article 46(f) of Directive 2001/83 to require manufacturing authorisation holders to verify that any excipients used in a product are made according to appropriate GMP standards. The Falsified Medicines Directive also committed the EC to publish Guidelines on the formalised risk assessment for ascertaining the appropriate GMP for excipients of medicinal products for human use (EC Guidelines), which were published on March 19, 2015. In 2018, PIC/S incorporated the same provisions for formalized risk assessment into a publication of the same name, extending the provisions to have global applicability.
To comply with the EC Guidelines and PIC/S publication, each excipient used in a drug product must be assessed for the risks that it poses to the product’s quality, safety, and purity. From these risks, the manufacturing authorization holder must determine the quality standards required for the manufacture of the excipient. The manufacturing authorization holder should communicate these quality standards to the excipient supplier and seek the supplier’s agreement. Any confirmed gaps in quality standards determined by the assessment must be appropriately mitigated, which may require the manufacturing authorization holder to take action on its own or through collaboration with the supplier. To ensure that risk assessments remain effective during the excipient’s lifecycle, the manufacturing authorization holder must incorporate a periodic review into the excipient risk assessment process.
Risk assessment for elemental impurity (ICH Q3D)
The product risk assessment would be focused on assessing the levels of elemental impurities in a drug product in relation to the PDEs presented in this guidance. The risk assessment process can be described in three steps:
- Identify known and potential sources of elemental impurities that may find their way into the drug product;
- Evaluate the presence of a particular elemental impurity in the drug product by determining the observed or predicted level of the impurity and comparing it with the established PDE;
- Summarize and document the risk assessment. Identify if controls built into the process are sufficient or identify additional controls to be considered to limit elemental impurities in the drug product.
In many cases, the steps are considered simultaneously. The outcome of the risk assessment may be the result of iterations to develop a final approach to ensure the potential elemental impurities do not exceed the PDE.
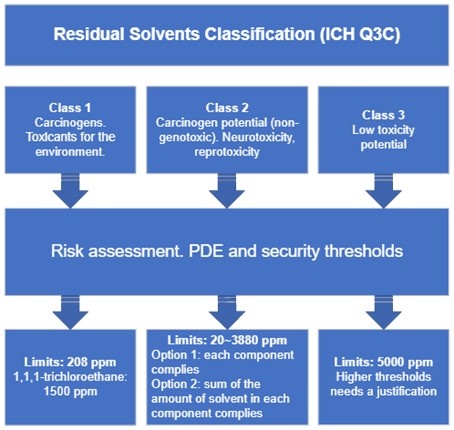
Risk assessment for residual solvents (ICH Q3C)
The product risk assessment would be focused on assessing the levels of residual solvents in a drug product in relation to the PDEs presented in this guidance. The risk assessment process can be described in three steps:
- Identify known and potential sources of residual solvents that may find their way into the drug product, then classify those solvents;
- Establish exposure limits depending on the solvent class;
- Identify if controls built into the process are sufficient or identify additional controls to be considered to limit elemental impurities in the drug product.
Risk assessment on the potential presence of N-nitrosamines
The aim of the risk assessment is:
- the identification of the potential sources of N-Nitrosamines and the ways of formation considering API and/or drug product manufacturing processes, as well as during shelf-life;
- the evaluation of the risk of the presence of N-Nitrosamines in the API and/or final drug product;
- the identification of the need for confirmatory testing in case of a potential risk of having N-nitrosamines inside API and/or final drug product is assessed.
Risk assessment in qualification/ validation
As part of a QRM system, decisions on the scope and extent of qualification and validation should be based on a justified and documented risk assessment of the facilities, equipment, utilities, and processes.
In equipment qualification, risk assessment may be carried out:
- To classify devices or systems into risk classes, then determine the scope and extent of the necessary qualification measures;
- To assess quality-relevant aspects and, if necessary, to define risk-minimizing measures in the URS and the scope and extent of each qualification phase.
In process validation, risk assessment may be carried out:
- To determine the number of batches manufactured and the number of samples taken;
- To identify process parameters and quality attributes as being critical or non-critical.
In transport verification, a risk assessment should be performed to consider the impact of variables in the transportation process other than those conditions which are continuously controlled or monitored, e.g. delays during transportation, failure of monitoring devices, topping up liquid nitrogen, product susceptibility, and any other relevant factors.
In qualification of utilities, a risk assessment should be carried out where there may be direct contact with the product, e.g. heating, ventilation, and air-conditioning (HVAC) systems, or indirect contact such as through heat exchangers to mitigate any risks of failure.
In cleaning validation, a risk assessment should be carried out:
- To define and justify the limits for the carryover of product residues;
- To determine an appropriate number of times running the cleaning procedure in order to prove that the cleaning method is validated;
- To select the worst-case product and determine the impact of new products on the site assessed;
- To determine and justify the sampling points.
Quality risk management should be used to evaluate planned changes to determine the potential impact on product quality, pharmaceutical quality systems, documentation, validation, regulatory status, calibration, maintenance, and any other system to avoid unintended consequences and to plan for any necessary process validation, verification or requalification efforts.

Reference
- ICH guideline Q10 on pharmaceutical quality system
- EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines – Part I
- 2015/C 95/02, Guidelines of 19 March 2015 on the formalised risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal products for human use
- PDA & IPEC Technical Report No. 54-6, Formalized Risk Assessment for Excipients
- European Commission. Guidelines of 19 March 2015 on the Formalised Risk Assessment for Ascertaining the Appropriate Good Manufacturing Practice for Excipients of Medicinal Products for Human Use. European Commission: Brussels, 2015
- ICHQ3C Impurities: Guideline for Residual Solvents
- ICHQ3D Guideline for Elemental Impurities
- Procedure number: EMEA/H/A-5(3)/1490, Nitrosamine impurities in human medicinal products
- EudraLex Volume 4, EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation



You must be logged in to post a comment.