
Một số ứng dụng quan trọng của Quản lý rủi ro chất lượng (QLRR) trong thực hành sản xuất thuốc tốt (GMP)
Quản lý rủi ro chất lượng đang dần trở thành một bộ phận không thể tách rời của một hệ thống chất lượng hiệu quả. Nó có thể cung cấp một cách tiếp cận chủ động để xác định, đánh giá một cách khoa học và kiểm soát các rủi ro tiềm ẩn đối với chất lượng cũng như hoạt động kinh doanh. Đồng thời, QLRR là một công cụ tốt để quản lý tri thức và tạo điều kiện cải tiến liên tục. Việc áp dụng QLRR rất đa dạng và tùy vào từng tổ chức mà có sự khác biệt, tuy nhiên, có một số rủi ro là yêu cầu bắt buộc trong GMP.
Đánh giá rủi ro trong nhiễm chéo
Quy trình QLRR nhằm mục đích đánh giá và kiểm soát các rủi ro nhiễm chéo. Việc đánh giá rủi ro thường được áp dụng cho từng phòng hoặc từng khu vực của kho, sản xuất, đóng gói dựa trên các con đường sau:
- Nhiễm chéo do nhầm lẫn
- Nhiễm chéo do sản phẩm còn sót lại tiếp xúc với các bề mặt
- Nhiễm chéo cơ học
- Nhiễm chéo qua không khí
Chính thức hóa việc đánh giá rủi ro nhằm xác định tiêu chuẩn GMP phù hợp đối với tá dược của các sản phẩm dùng trên người.
Tá dược đưa vào công thức sản phẩm thuốc vì nhiều lý do: để hỗ trợ công thức, đóng vai trò là thành phần chức năng hoặc không chức năng hoặc để cho phép thuốc được giải phóng khỏi sản phẩm thuốc theo một cách cụ thể nào đó. Cơ sở lý luận cho việc sử dụng các tá dược này không theo quan điểm phục vụ mục đích y tế mà mở rộng ra bao gồm luôn việc duy trì sự an toàn và chức năng của sản phẩm, thậm chí trong suốt quá trình bảo quản. Trong khi các quy định về GMP nêu rõ những gì cần thiết đối với hoạt chất, ngược lại chỉ có một khung cơ bản để xác định GMP thích hợp cho tá dược dựa vào đánh giá rủi ro.
Để ghi nhận mức độ quan trọng của tá dược đối với một thuốc, chỉ thị về Thuốc giả của Ủy ban Châu Âu (EC) (2011/62/EU) đã sửa đổi điều 46 (f) của chỉ thị 2001/83 để yêu cầu chủ sở hữu giấy phép sản xuất xác minh rằng bất kỳ tá dược sử dụng trong sản phẩm đều được sản xuất theo tiêu chuẩn GMP thích hợp. Chỉ thị về Thuốc giả cũng cam kết EC sẽ xuất bản Hướng dẫn về Chính thức hóa việc đánh giá rủi ro nhằm xác định tiêu chuẩn GMP phù hợp đối với tá dược của các sản phẩm dùng trên người (Hướng dẫn của EC), được xuất bản vào ngày 19 tháng 3 năm 2015. Năm 2018, tổ chức PIC/S (Pharmaceutical Inspection Co-operation Scheme) đã hợp nhất các điều khoản tương tự về chính thức hóa việc đánh giá rủi ro để thành một ấn phẩm cùng tên, mở rộng các điều khoản để có khả năng áp dụng toàn cầu.
Để tuân thủ hướng dẫn của EC và công bố của PIC/S, mỗi tá dược dùng trong thuốc phải được đánh giá các rủi ro mà nó có thể gây ra đối với chất lượng, độ an toàn và độ tinh khiết của sản phẩm thuốc. Từ những rủi ro này, người được cấp phép sản xuất phải xác định các tiêu chuẩn chất lượng cần thiết cho việc sản xuất tá dược. Người được ủy quyền sản xuất phải thông tin cho nhà cung cấp tá dược về các tiêu chuẩn chất lượng này và tìm kiếm sự đồng ý từ họ. Bất kì một lỗ hổng nào đã được xác nhận trong tiêu chuẩn chất lượng thông qua đánh giá phải được khắc phục một cách thích hợp, tùy vào trường hợp mà việc khắc phục này có thể thực hiện bằng việc phối hợp với nhà cung cấp tá dược hoặc bởi chính phía sở hữu giấy phép sản xuất. Để đảm bảo rằng các đánh giá rủi ro vẫn có hiệu quả trong suốt vòng đời của tá dược, người sở hữu giấy phép sản xuất phải kết hợp việc xem xét định kỳ vào quá trình đánh giá rủi ro tá dược.
Đánh giá rủi ro đối với tạp kim loại nặng (ICH Q3D)
Đánh giá rủi ro sản phẩm sẽ tập trung vào đánh giá mức độ tạp nguyên tố trong sản phẩm thuốc liên quan đến Liều lượng cho phép hằng ngày (Permitted Daily Dose – PDE) được trình bày trong hướng dẫn này. Quá trình đánh giá rủi ro có thể được mô tả theo ba bước:
- Xác định các nguồn tạp nguyên tố đã biết cũng như tiềm ẩn có thể xâm nhập vào sản phẩm thuốc.
- Đánh giá sự hiện diện của một tạp nguyên tố cụ thể trong sản phẩm thuốc bằng cách xác định mức độ tạp chất quan sát được hoặc dự đoán được, sau đó so sánh với PDE đã thiết lập.
- Tổng hợp và lập hồ sơ đánh giá rủi ro. Xác định xem các biện pháp kiểm soát được tích hợp trong quá trình đã đủ chưa hoặc xác định các biện pháp kiểm soát bổ sung để hạn chế tạp nguyên tố trong sản phẩm thuốc.
Trong nhiều trường hợp, các bước được coi là đồng thời. Kết quả của việc đánh giá rủi ro có thể là kết quả của các lần lặp lại để phát triển một cách tiếp cận cuối cùng nhằm đảm bảo các tạp nguyên tố tiềm ẩn không vượt quá PDE.
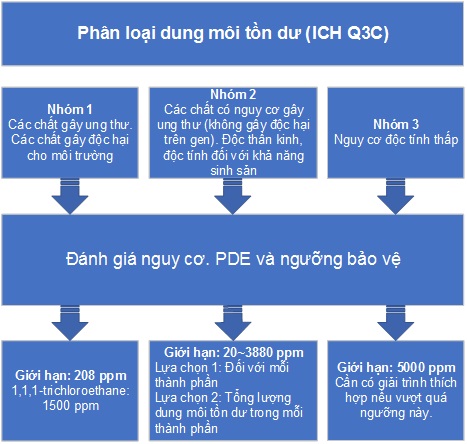
Đánh giá rủi ro đối với dung môi tồn dư (ICH Q3C)
Đánh giá rủi ro sản phẩm sẽ tập trung vào việc đánh giá mức độ dung môi tồn dư trong sản phẩm thuốc liên quan đến PDE được trình bày trong hướng dẫn này. Quá trình đánh giá rủi ro có thể được mô tả theo ba bước:
- Xác định các nguồn dung môi tồn dư tiềm ẩn và đã biết trước đó. Là các nguồn dung môi mà có thể tồn tại trong thuốc do một con đường nào đó, tiếp theo, phân loại các dung môi này;
- Thiết lập giới hạn phơi nhiễm phụ thuộc vào loại dung môi;
- Nhận biết nếu các biện pháp kiểm soát được xây dựng trong quy trình là đủ hoặc xác định các biện pháp kiểm soát bổ sung được xem xét để hạn chế các tạp nguyên tố trong sản phẩm thuốc.
Đánh giá rủi ro về sự hiện diện tiềm ẩn của N-Nitrosamine
Mục đích của việc đánh giá rủi ro là:
- Xác định các nguồn có nguy cơ của các N-Nitrosamine và những con đường tạo thành hoạt chất và/hoặc các quy trình sản xuất ra thuốc, cũng như trong suốt thời gian lưu hành của thuốc;
- Đánh giá nguy cơ tồn tại của các N-Nitrosamine trong hoạt chất và/hoặc sản phẩm cuối cùng;
- Xác định sự cần thiết của thử nghiệm xác nhận trong trường hợp có nguy cơ tiềm ẩn tồn tại các N-Nitrosamine bên trong API và/hoặc đánh giá sản phẩm cuối cùng.
Đánh giá rủi ro trong đánh giá/thẩm định
Như một phần của hệ thống QLRR, các quyết định về phạm vi và mức độ đánh giá và xác nhận phải dựa trên đánh giá rủi ro được chứng minh và lập thành văn bản đối với cơ sở vật chất, thiết bị, tiện ích và quy trình.
Đối với đánh giá thiết bị, việc đánh giá rủi ro có thể được thực hiện như sau:
- Phân chia các thiết bị và hệ thống vào các nhóm nguy cơ, sau đó xác định phạm vi và mức độ cần thiết của các biện pháp đánh giá;
- Đánh giá các khía cạnh liên quan chất lượng và nếu cần thiết thì xác định phương pháp giảm thiểu rủi ro trong tiêu chuẩn yêu cầu của người dùng (User requirement specification – URS) và phạm vi cũng như mức độ của mỗi giai đoạn đánh giá.
Đối với thẩm định quy trình, đánh giá rủi ro có thể được thực hiện như sau:
- Xác định số lô sản xuất và số mẫu cần lấy;
- Xác định các thông số quy trình và thuộc tính chất lượng là quan trọng hay không quan trọng.
Đối với xác minh việc vận chuyển, cần thực hiện đánh giá rủi ro để xem xét tác động của các biến số ngoài những điều kiện được kiểm soát hoặc giám sát liên tục trong quá trình vận chuyển, ví dụ: chậm trễ trong quá trình vận chuyển, thiết bị giám sát hỏng, tích tụ ni-tơ lỏng, sự nhạy cảm của sản phẩm và bất kỳ yếu tố liên quan nào khác.
Đối với đánh giá các hệ thống phụ trợ, cần thực hiện đánh giá rủi ro làm giảm nguy cơ hư hỏng khi chúng có thể tiếp xúc trực tiếp với sản phẩm, ví dụ: hệ thống nhiệt, thông gió và điều hòa không khí (HVAC) hoặc tiếp xúc gián tiếp như qua bộ trao đổi nhiệt.
Đối với thẩm định vệ sinh, việc đánh giá rủi ro nên được thực hiện như sau:
- Xác định và đánh giá giới hạn mang qua của sản phẩm tồn dư;
- Đưa ra số lần thực hiện quy trình vệ sinh thích hợp để chứng minh phương pháp vệ sinh đã được thẩm định;
- Lựa chọn sản phẩm nhiều nguy cơ nhất và quyết định ảnh hưởng của sản phẩm mới lên chuyền sản xuất;
- Đưa ra và giải thích rõ các vị trí lấy mẫu.
QLRR nên được áp dụng để đánh giá các thay đổi theo kế hoạch nhằm xác định tác động tiềm tàng đối với chất lượng sản phẩm, hệ thống chất lượng dược phẩm, tài liệu, thẩm định, tình trạng pháp lý, hiệu chuẩn, bảo trì và bất kì hệ thống nào khác để tránh hậu quả không mong muốn; nhằm lập kế hoạch cho bất kỳ thẩm định quy trình cần thiết nào, xác minh hoặc các nỗ lực yêu cầu.

Tài liệu tham khảo
- ICH guideline Q10 on pharmaceutical quality system
- EudraLex – Volume 4 – Good Manufacturing Practice (GMP) guidelines – Part I
- 2015/C 95/02, Guidelines of 19 March 2015 on the formalised risk assessment for ascertaining the appropriate good manufacturing practice for excipients of medicinal products for human use
- PDA & IPEC Technical Report No. 54-6, Formalized Risk Assessment for Excipients
- European Commission. Guidelines of 19 March 2015 on the Formalised Risk Assessment for Ascertaining the Appropriate Good Manufacturing Practice for Excipients of Medicinal Products for Human Use. European Commission: Brussels, 2015
- ICHQ3C Impurities: Guideline for Residual Solvents
- ICHQ3D Guideline for Elemental Impurities
- Procedure number: EMEA/H/A-5(3)/1490, Nitrosamine impurities in human medicinal products
- EudraLex Volume 4, EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation



You must be logged in to post a comment.