
Hệ thống phê duyệt thuốc mới của Châu Âu bao gồm thủ tục ủy quyền trên toàn Liên minh Châu Âu (EU) (được gọi là quy trình tập trung) cùng với các thủ tục quốc gia dựa trên các Quốc gia thành viên EU khác nhau làm việc cùng nhau và công nhận các đánh giá của nhau (cái gọi là phân cấp và các thủ tục thừa nhận lẫn nhau). Đây là một hệ thống đã phát triển trong hơn nửa thế kỷ qua từ một hệ thống với các hệ thống quốc gia hoàn toàn riêng biệt thành một hệ thống mà các nước EU hiện đang khai thác chuyên môn khoa học và pháp lý của họ để hài hòa và cải thiện việc đánh giá thuốc trên toàn châu Âu. Ngày nay, thủ tục quốc gia thuần túy hiếm khi được người nộp đơn sử dụng và chỉ khi họ xin phép tiếp thị ở một Quốc gia Thành viên. Mặc dù các quy trình khác nhau có thể gây ra các vấn đề phức tạp, nhưng chúng đã đơn giản hóa quy trình cấp phép giữa các Quốc gia Thành viên, giảm thời gian để các loại thuốc mới được cấp phép lưu hành và cải thiện khả năng tiếp cận của bệnh nhân với các loại thuốc mới.
Hệ thống phê duyệt thuốc hiện tại của Châu Âu bao gồm thủ tục ủy quyền tập trung cũng như thủ tục ủy quyền quốc gia dựa trên sự ủy quyền đồng thời tại nhiều Quốc gia Thành viên của Liên minh Châu Âu (EU) và sự công nhận lẫn nhau về ủy quyền tiếp thị. Ngoài ra, có những loại thuốc được ủy quyền tại các Quốc gia Thành viên theo thủ tục quốc gia thuần túy. Quy trình tập trung và Cơ quan Dược phẩm Châu Âu, cơ quan quản lý quy trình, đều đã hoạt động từ năm 1995. Bài báo này mô tả lịch sử của hệ thống phê duyệt và sự hài hòa đã xảy ra trong nửa thế kỷ qua và cung cấp một cái nhìn tổng quan về cách thức thuốc được chấp thuận ở EU ngày nay.
Lịch sử của hệ thống quản lý dược phẩm ở Châu Âu
Mặc dù nhiều nước châu Âu từ lâu đã có luật quy định việc sử dụng các loại thuốc khác nhau, quy định về dược phẩm hiện đại ở châu Âu có thể được coi là bắt đầu từ những năm 1960 và đã không còn đứng yên kể từ đó. Sau thảm kịch thalidomide, việc tăng cường kiểm soát pháp lý đối với dược phẩm đã được tăng cường, với các cơ quan quản lý được thành lập trên khắp Châu Âu để phê duyệt thuốc và các Quốc gia Thành viên đang nỗ lực hài hòa hóa Châu Âu, dẫn đến Chỉ thị dược phẩm đầu tiên vào năm 1965 (Chỉ thị của Hội đồng 65/65/EEC). Chỉ thị yêu cầu tất cả các loại thuốc phải có giấy phép lưu hành và cũng nhằm mục đích hài hòa các tiêu chuẩn để phê duyệt thuốc ở Châu Âu. Ngoài ra, luật này khuyến khích việc thành lập một thị trường dược phẩm duy nhất ở EU vào thời điểm mà mỗi quốc gia đều có các thủ tục phê duyệt riêng biệt của mình, điều đó có nghĩa là các công ty phải nộp các đơn đăng ký riêng biệt để được phê duyệt thuốc ở mỗi quốc gia.
Năm 1975, hai Chỉ thị của Hội đồng đã được đưa ra, chỉ thị thứ nhất (Chỉ thị của Hội đồng 75/318/liên quan đến việc thử nghiệm các loại thuốc được yêu cầu thực hiện bởi các công ty xin phép lưu hành, và chỉ thị thứ hai (Chỉ thị của Hội đồng 75/319/EEC, 1975) thiết lập thủ tục cấp phép lưu hành với mục đích thúc đẩy sự di chuyển miễn phí của thuốc. Quy trình này dựa trên sự công nhận lẫn nhau của các đánh giá quốc gia, theo đó một công ty có thể xin phép lưu hành thuốc ở một Quốc gia Thành viên trên cơ sở giấy phép tiếp thị hiện có ở một Quốc gia Thành viên khác. Chỉ thị cũng thành lập một ủy ban cố vấn cho Ủy ban châu Âu được gọi là Ủy ban về các sản phẩm thuốc độc quyền (CPMP) để giúp các Quốc gia Thành viên EU áp dụng quan điểm chung về quyết định cấp phép lưu hành. Tuy nhiên, các ý kiến của CPMP không có tính ràng buộc và hệ thống này đã bị chỉ trích là chậm chạp, quan liêu và kém hiệu quả, trong đó các Quốc gia Thành viên không công nhận các đánh giá của nhau và hầu như mọi trường hợp đều tìm kiếm sự phân xử từ4CPMP được gọi là ‘thủ tục CPMP’ và sau đó đã được đơn giản hóa và trở thành ‘thủ tục cấp phép đa bang’. Tuy nhiên, quy trình này, mặc dù đã được cải tiến, nhưng vẫn bị nhiều người coi là không hiệu quả và ít được ngành công nghiệp sử dụng.5 Năm 1985, dự án thị trường đơn lẻ được khởi động, trong đó có kế hoạch thành lập Cơ quan Thuốc Châu Âu. Năm 1986, một thủ tục mới để cấp phép thuốc được gọi là ‘thủ tục hòa nhạc’ đã được giới thiệu. Thủ tục này là bắt buộc đối với các loại thuốc công nghệ sinh học, cần có ý kiến cấp phép trên toàn cộng đồng của CPMP đối với các loại thuốc này trước khi có thể cấp phép lưu hành ở bất kỳ Quốc gia Thành viên nào. Tuy nhiên, ý kiến này một lần nữa không có giá trị ràng buộc đối với các Quốc gia Thành viên và các Quốc gia Thành viên vẫn có thể chấp thuận hoặc từ chối đơn đăng ký mà không cần tham khảo ý kiến đó.
Một bước quan trọng trong quá trình hài hòa đã được thực hiện vào năm 1993 với Quy định của Hội đồng (EEC) 2309/93, thành lập Cơ quan Châu Âu về Đánh giá Sản phẩm Thuốc, nay được gọi là Cơ quan Dược phẩm Châu Âu. Ngoài ra, quy trình tập trung đã được sửa đổi và trở thành thủ tục tập trung. Quy định, có hiệu lực vào năm 1995, cũng tái thiết lập CPMP thành CPMP ‘mới’ để đưa ra ý kiến của Cơ quan về việc cấp phép tiếp thị theo quy trình tập trung, hiện đã dẫn đến các quyết định ràng buộc về mặt pháp lý của Ủy ban. CPMP sau đó được đổi tên thành Ủy ban Sản phẩm Thuốc dùng cho Con người (CHMP).
Do phạm vi bắt buộc của quy trình tập trung mới chỉ giới hạn trong các loại thuốc công nghệ sinh học, nên nó đã thay thế các quy trình quốc gia hiện có đối với các loại thuốc này. Khái niệm công nhận lẫn nhau đối với các loại thuốc khác vẫn được duy trì và được đưa vào luật dược Châu Âu vào năm 1993 (Council Directive 93/39/EEC, 1993).
Đến năm 1995, một hệ thống phê duyệt thuốc hài hòa của Châu Âu do đó đã xuất hiện bao gồm một thủ tục dựa trên sự thừa nhận lẫn nhau về việc cấp phép lưu hành của các Quốc gia Thành viên và mặt khác là một thủ tục cung cấp ý kiến cấp phép cho toàn cộng đồng. Thủ tục thừa nhận lẫn nhau có hai tiền thân: thứ nhất, thủ tục CPMP hoạt động từ năm 1976 đến năm 1985; sau đó là thủ tục cấp phép đa bang hoạt động từ năm 1985 đến năm 1995, mà vào năm 1995, được gọi là thủ tục công nhận lẫn nhau. Thủ tục cung cấp ý kiến cấp phép rộng rãi của cộng đồng, thủ tục tập trung, được phát triển từ thủ tục hòa nhạc hoạt động từ năm 1986 đến năm 1995. Trong khi các thủ tục ban đầu bị cản trở do CPMP thiếu ý kiến ràng buộc, đến năm 1995, quy trình này không còn nữa và quy định dược phẩm ở Châu Âu đã trở nên hài hòa hơn và hiệu quả hơn. Sau năm 1995, những thay đổi bổ sung đã được thực hiện đối với hệ thống phê duyệt của Châu Âu này để tăng cường hơn nữa. Chúng bao gồm giới thiệu vào năm 2005, sự ra đời của một thủ tục mới được gọi là thủ tục phi tập trung nhằm tránh khả năng xảy ra tranh chấp mà theo thời gian đã được xác định là một vấn đề với thủ tục thừa nhận lẫn nhau vì các Quốc gia Thành viên trong đó yêu cầu sự chấp thuận đã không tham gia đủ sớm đánh giá.

Hệ thống EU hiện tại
Thủ tục tập trung
điểm của quy trình tập trung là nó yêu cầu một ứng dụng duy nhất, nếu thành công, sẽ dẫn đến một ủy quyền tiếp thị duy nhất với cùng thông tin sản phẩm có sẵn bằng tất cả các ngôn ngữ của EU và hợp lệ ở tất cả các nước EU, cũng như Iceland, Liechtenstein và Na Uy. Đánh giá khoa học về đơn xin cấp phép lưu hành do CHMP thực hiện. Quy trình đánh giá khoa học bao gồm các giai đoạn đánh giá tích cực xen kẽ và các khoảng thời gian đồng hồ dừng lại để người nộp đơn có thời gian giải quyết mọi vấn đề được xác định trong quá trình đánh giá. Tổng cộng, thời gian của quy trình lên đến 210 ngày “hoạt động” trước khi CHMP đưa ra ý kiến.
Sau khi một ý kiến đã được đưa ra, nó sẽ được chuyển đến Ủy ban Châu Âu, sau đó có 67 ngày để đưa ra quyết định ràng buộc về mặt pháp lý đối với việc cấp phép tiếp thị. Thời gian phê duyệt trung bình cho các loại thuốc trong năm 2012 được EMA phê duyệt là 14,8 tháng. Khi đã được cấp phép lưu hành, người nộp đơn có thể bắt đầu tiếp thị thuốc ở bất kỳ Quốc gia Thành viên EU nào mà mình lựa chọn. Tuy nhiên, trên thực tế, trước khi một loại thuốc được bán trên thị trường, nó sẽ phải trải qua các cuộc đàm phán về giá cả và xem xét tính hiệu quả về chi phí của nó. Việc này được thực hiện ở cấp quốc gia bởi các Quốc gia Thành viên để xác định các tiêu chí bồi hoàn.
Ban đầu, quy trình tập trung chỉ là bắt buộc đối với các loại thuốc công nghệ sinh học, như trường hợp của quy trình hòa nhạc trước đó. Tuy nhiên, theo thời gian, phạm vi bắt buộc của thủ tục tập trung đã dần được mở rộng và đến năm 2005, nó bao gồm các loại thuốc dành cho trẻ mồ côi (thuốc chữa các bệnh hiếm gặp) cũng như các loại thuốc dành cho người có chứa một hoạt chất mới (trước đây không được phép trong Liên minh trước năm 20 Tháng 11 năm 2005) và được dùng để điều trị bệnh AIDS, ung thư, rối loạn sinh tế bào thần kinh, bệnh tiểu đường, rối loạn chức năng miễn dịch tự động và miễn dịch khác, và các bệnh do vi rút. Năm 2009, quy trình tập trung cũng trở thành bắt buộc đối với các loại thuốc trị liệu tiên tiến. Quy trình tập trung cũng là tùy chọn đối với các loại thuốc khác có chứa hoạt chất mới không được Liên minh ủy quyền trước ngày 20 tháng 11 năm 2005 và đối với các sản phẩm được coi là một phương pháp điều trị, khoa học hoặc đổi mới kỹ thuật quan trọng hoặc cho toàn EU ủy quyền được coi là vì lợi ích của sức khỏe cộng đồng (Hình 1).
Loại thuốc đầu tiên được ủy quyền theo quy trình tập trung là thuốc điều trị hiếm muộn Gonal-F vào tháng 10 năm 1995. Hiện EMA nhận được khoảng
100 đơn đăng ký mỗi năm (Hình 2), trong đó, khoảng 10% không đưa ra ý kiến nhưng bị rút lại, và khoảng 5% dẫn đến ý kiến tiêu cực (Hình 3). Kể từ khi cơ quan được thành lập vào năm 1995, hơn 700 loại thuốc chữa bệnh cho người đã được phê duyệt theo quy trình tập trung. Trong những năm đầu, chỉ các sản phẩm cải tiến mới được phê duyệt thông qua quy trình tập trung nhưng khi tính độc quyền dữ liệu cho các sản phẩm đầu tiên được phê duyệt bắt đầu hết hạn, các thuốc generic cũng được phê duyệt tập trung. Số lượng đơn đăng ký thuốc generic sử dụng quy trình tập trung đã tăng lên trong những năm qua, đạt đỉnh vào năm 2010 với khoảng 50% tất cả các ứng dụng là thuốc gốc (Hình 4). Ngày nay, hầu hết các loại thuốc có chứa hoạt chất mới đều được phê duyệt bằng cách sử dụng quy trình tập trung.
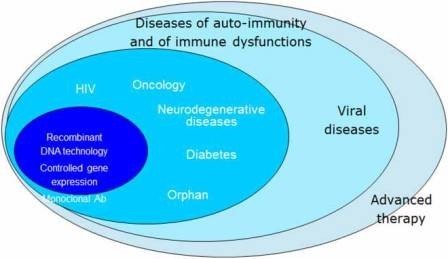
Hình 1: Phạm vi bắt buộc của thủ tục tập trung.
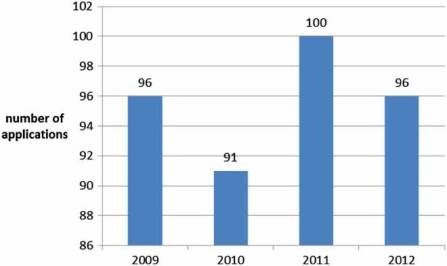
Hình 2: Số lượng đơn đăng ký nhận được hàng năm bởi EMA (2009–2012).
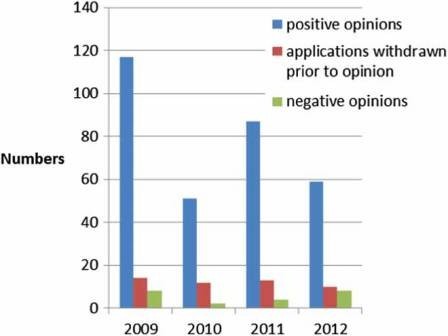
Hình 3: Kết quả đánh giá (2009–2012).
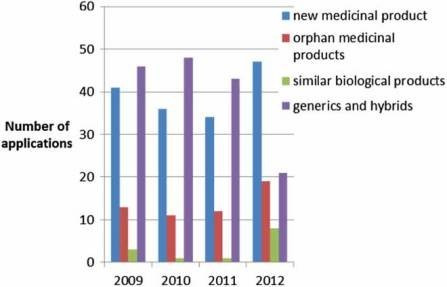
Hình 4: Loại đơn được nhận bởi EMA (2010-2012).
Thủ tục công nhận lẫn nhau
Thủ tục công nhận lẫn nhau đã được áp dụng từ năm 1995 và phát triển từ thủ tục cấp phép đa quốc gia. Người nộp đơn ban đầu phải nhận được sự chấp thuận của quốc gia tại một Quốc gia Thành viên EU, được gọi là ‘Quốc gia Thành viên tham chiếu’ và sau đó tìm kiếm sự chấp thuận cho thuốc ở một quốc gia khác, được gọi là ‘Quốc gia Thành viên có liên quan’ trong bước thứ hai dựa trên đánh giá được thực hiện tại quốc gia thành viên tham chiếu. Quy trình này có những điểm khác biệt đáng kể so với quy trình cấp phép đa quốc gia trước đây, đáng chú ý là yêu cầu rằng các bất đồng giữa các Quốc gia Thành viên giờ đây phải được giải quyết ở cấp độ EU. Các bất đồng được xử lý bởi Nhóm điều phối các Thủ tục Công nhận và Phân cấp lẫn nhau – Con người (CMDh), một cơ quan đại diện cho các Quốc gia Thành viên, chịu trách nhiệm về bất kỳ câu hỏi nào ở hai hoặc nhiều Quốc gia Thành viên liên quan đến việc cấp phép lưu hành một sản phẩm thuốc đã được phê duyệt thông qua công nhận lẫn nhau hoặc thủ tục phi tập trung.
Nếu có bất đồng giữa các Quốc gia Thành viên với lý do tiềm ẩn nguy cơ nghiêm trọng đối với sức khỏe cộng đồng, CMDh sẽ xem xét vấn đề này để đạt được thỏa thuận trong vòng 60 ngày. Nếu điều này là không thể, thủ tục được chuyển đến CHMP trong một thủ tục được gọi là chuyển tuyến. CHMP sau đó sẽ thay mặt EU thực hiện đánh giá khoa học về loại thuốc có liên quan.
Ngược lại với thủ tục trước đây, kết quả của CHMP ràng buộc đối với các Quốc gia thành viên có liên quan sau khi nó được Ủy ban Châu Âu thông qua. Thời hạn để CHMP đánh giá là 60 ngày.
Kể từ khi thủ tục phi tập trung ra đời, thủ tục công nhận lẫn nhau được sử dụng để mở rộng ủy quyền tiếp thị hiện có cho các quốc gia khác.

Thủ tục phi tập trung
Trong thủ tục phi tập trung, người nộp đơn chọn một quốc gia làm Quốc gia Thành viên tham chiếu khi nộp đơn xin cấp phép lưu hành. Sau đó, Quốc gia Thành viên tham chiếu được chọn sẽ chuẩn bị trước một bản dự thảo báo cáo đánh giá được đệ trình cho các Quốc gia Thành viên khác, nơi họ đang tìm kiếm sự chấp thuận để họ xem xét và phê duyệt đồng thời. Khi cho phép các Quốc gia Thành viên khác tiếp cận đánh giá này ở giai đoạn đầu, mọi vấn đề và mối quan tâm có thể được giải quyết nhanh chóng mà không bị chậm trễ, điều này đôi khi được biết là xảy ra với thủ tục công nhận lẫn nhau. So với thủ tục thừa nhận lẫn nhau, thủ tục phi tập trung có ưu điểm là giấy phép lưu hành ở tất cả các Quốc gia Thành viên đã chọn được nhận đồng thời, cho phép tiếp thị thuốc đồng thời và giảm gánh nặng hành chính và quản lý. Ngày nay, thủ tục phi tập trung chủ yếu được sử dụng cho ứng dụng cho thuốc gốc.
Đối với thủ tục thừa nhận lẫn nhau, các bất đồng được CMDh hoặc CHMP xử lý trong trường hợp không thể đạt được thỏa thuận ở cấp CMDh. Tóm lại, các thủ tục phê duyệt thuốc hiện tại ở Châu Âu là kết quả của nỗ lực hài hòa và cải thiện quy định về thuốc và đối với hầu hết các loại thuốc, đã thay thế các phê duyệt dựa trên sự cho phép hoàn toàn của quốc gia. Trong những năm qua, phạm vi của quy trình tập trung đã được mở rộng và ngày nay hầu hết các loại thuốc có chứa hoạt chất mới đều được chấp thuận sử dụng quy trình tập trung. Thủ tục công nhận lẫn nhau và phân cấp chủ yếu được sử dụng để mở rộng ủy quyền tiếp thị hiện có hoặc cho các loại thuốc thông thường. Nửa thế kỷ hài hòa đã dẫn đến một hệ thống được đơn giản hóa, cải thiện khả năng tiếp cận thuốc bằng cách giảm thời gian cho các loại thuốc mới để có được giấy phép lưu hành.
Cre: Inga Abed/European Medicines Agency London, UK
Info: Inga.abed@ema.europa.eu



You must be logged in to post a comment.