
1. Giới thiệu
Tạp chất được coi là bất kỳ thành phần nào của dược chất mà thành phần hóa học của nó được xác định không phải là dược chất và ngoài ra, đối với sản phẩm thuốc, bất kỳ thành phần nào không phải là thành phần công thức cũng được xem là tạp chất [1]. Hồ sơ tạp chất là cơ sở để xác định, đảm bảo chất lượng, an toàn và hiệu quả của các dược chất và các sản phẩm thuốc.
Thiết lập hồ sơ tạp chất của dược chất (hoạt chất) là cơ sở cho việc thiết lập hồ sơ tạp chất của các sản phẩm thuốc, tuy nhiên, quá trình này cũng đồng thời xem xét đến tá dược và quá trình bào chế. Bài viết này sẽ thảo luận về các hướng dẫn được xem xét trong việc phân loại các tạp chất, xác định nguồn tiềm năng, cách đánh giá, phương pháp phân tích có thể được sử dụng, xác định mức tạp chất và giới hạn đề xuất.
Tạp chất trong dược chất và sản phẩm thuốc có thể được phân loại như dưới đây theo các hướng dẫn hiện hành của ICH, FDA, EU.
a) Tạp hữu cơ
I. Tạp quá trình
II. Tạp phân hủy
III. Tạp đồng phân
b) Tạp độc tính lên gen
c) Tạp vô cơ
d) Dung môi tồn dư
e) Tạp đa hình
Quá trình phát triển của xác định hồ sơ tạp chất trong dược chất và sản phẩm thuốc qua các thời kỳ có thể được trình bày như Hình 1.
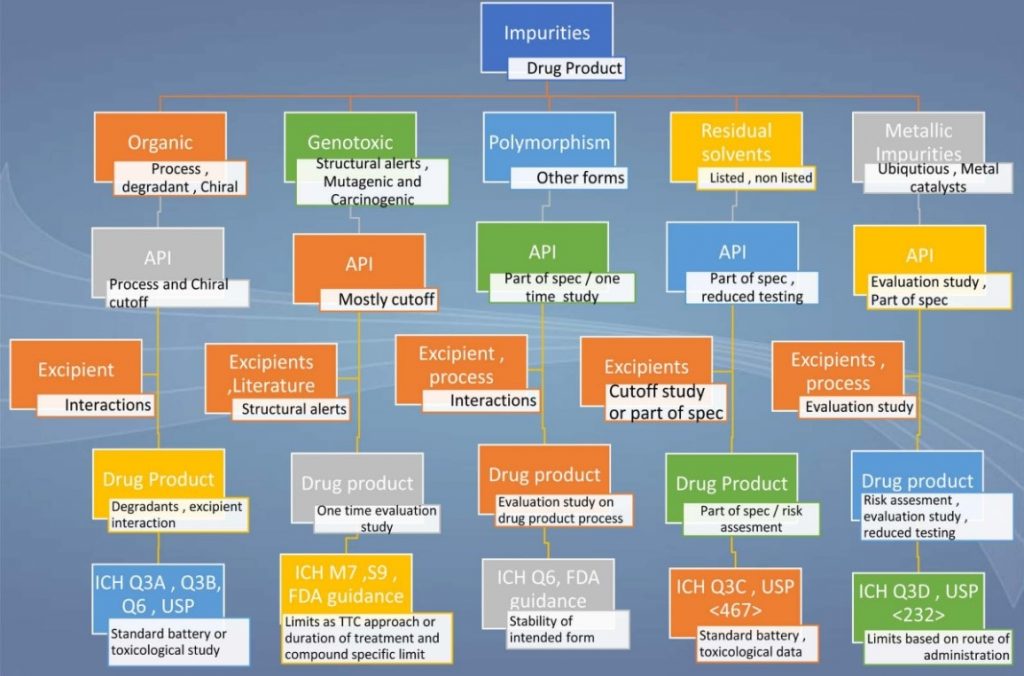
Một ảnh giới thiệu về hồ sơ tạp chất của dược chất và sản phẩm thuốc được minh họa trong Hình 2.
2. Tạp chất hữu cơ
Tổng quan về hồ sơ tạp chất hữu cơ được mô tả chi tiết ở Hình 3.
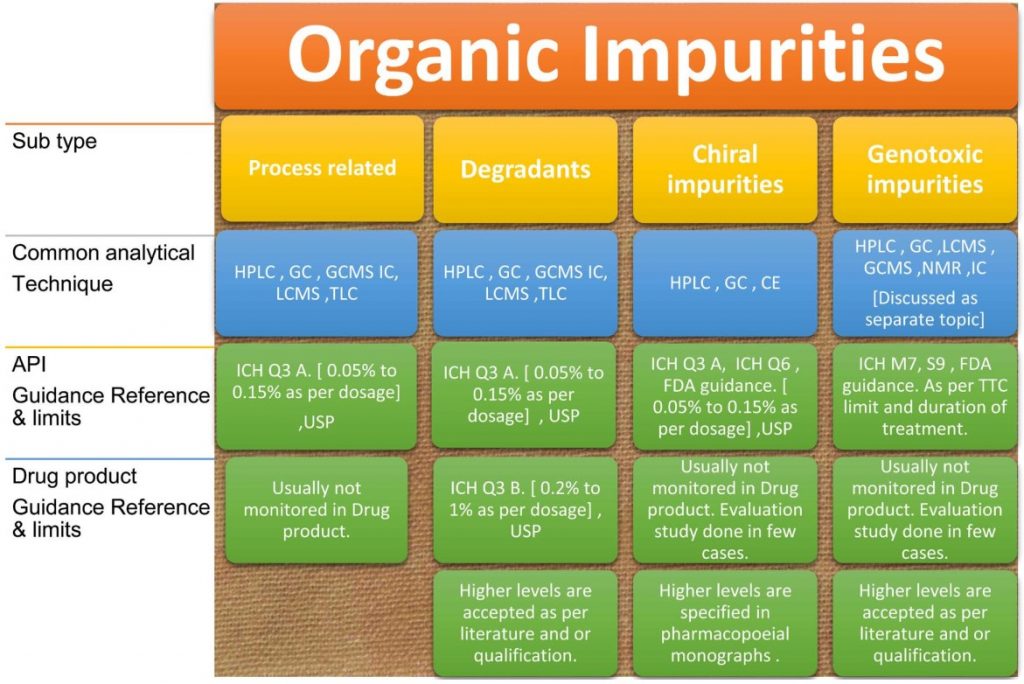
Tạp hữu cơ được phân loại rộng ra thành các loại:
1.1. Tạp liên quan đến quá trình
Tạp từ nguyên liệu ban đầu, sản phẩm phụ, sản phẩm trung gian, thuốc thử, phối tử, và chất xúc tác mà không gia tăng trong nghiên cứu độ ổn định và không hình thành trong quá trình nghiên cứu lão hóa điều kiện khắc nghiệt được xem như là tạp liên quan đến quá trình.
Tạp liên quan đến quá trình được hình thành trong quá trình tổng hợp, tinh khiết hóa và bảo quản của dược chất. Hầu hết các tạp này được mong đợi trong quá trình tổng hợp dựa theo hiểu biết về quá trình tổng hợp, loại hóa chất/thuốc thử được sử dụng, cũng như các tạp này được xác nhận bằng nghiên cứu độ ổn định [4] và nghiên cứu lão hóa. Việc lựa chọn các tạp chất nên được xác minh chéo hoặc thách thức bởi các nghiên cứu trong điều kiện khắc nghiệt [12].
Tạp chất trong tiêu chuẩn chất lượng của thành phẩm thuốc có thể được đề xuất dựa theo các tạp chất được xác định trong quá trình sản xuất công nghiệp để thiết lập việc kiểm soát quá trình tốt hơn. Tạp nào mà cao hơn ngưỡng đánh giá nên được xem xét trong tiêu chuẩn thành phẩm cùng với ngưỡng báo cáo và ngưỡng đánh giá dựa theo mức độ an toàn của các ngưỡng này.
Trong khi ở sản phẩm thuốc, việc chứng minh tính đặc hiệu (thường là sự phân tách ở phương pháp sắc ký) cho tạp liên quan đến quá trình cùng với tạp phân hủy là yêu cầu đối với phương pháp theo dõi độ ổn định nhưng không có yêu cầu cho việc báo cáo tạp chất liên quan đến quá trình (khi mà chúng được kiểm soát trong nguyên liệu được sử dụng) trừ khi điều này được đề cập trong chuyên luận dược điển tương ứng.
1.1.1. Kỹ thuật phân tích
Các kỹ thuật hiện đại để định lượng các tạp này là sắc ký lỏng hiệu năng cao (HPLC), sắc kí lỏng siêu hiệu năng (UPLC)/ sắc ký lỏng phân giải nhanh (PPLC), sắc kí khí (GC), sắc ký lỏng ghép khối phổ (LC-MS), sắc ký khí ghép khối phổ (GC-MS).
Giới hạn được thiết lập theo hướng dẫn của ICH Q3A “Tạp chất trong các dược chất mới”.
1.1.2. Tiêu chuẩn chất lượng cho dược chất [2]
Liều dung tối đa hằng ngày: ≤ 2 g/ngày: 0.15% hoặc 1.0 mg trên ngày dùng (lấy giá trị nào thấp hơn).
Liều dùng tối đa hằng ngày: > 2 g/ngày: 0.05%.
1.1.3. Tiêu chuẩn chất lượng sản phẩm thuốc
Không được xem xét để báo cáo trừ khi được đề cập trong chuyên luận riêng trong dược điển.
1.2. Tạp phân hủy/sản phẩm phân hủy
Nghiên cứu lão hóa điều kiện khắc nghiệt nên được thực hiện để xác định sản phẩm phân hủy tiềm tàng, đặc tính của sản phẩm phân hủy, độ ổn định của phân tử hoạt chất, để hiểu về con đường phân hủy của thuốc, tốc độ phân hủy, phát triển phương pháp phân tích theo dõi độ ổn định và để xác định hạn dùng.
Những tạp chất mà gia tăng trong nghiên cứu độ ổn định (Q1A) hay được tạo ra trong nghiên cứu lão hõa điều kiện khắc nghiệt được phân loại là tạp phân hủy/sản phẩm phân hủy.
Việc lựa chọn các điều kiện cho nghiên cứu lão hóa và mức độ lão hóa hướng đến được thực hiện theo các hướng dẫn thực hành hiện hành. Hướng dẫn ICH Q1B “Thử nghiệm độ ổn định trong điều kiện ánh sáng của dược chất mới và sản phẩm mới” được xem như là một tài liệu hướng dẫn về độ ổn định trong điều kiện ánh sáng [13]
1.2.1. Kỹ thuật phân tích
Các kỹ thuật sử dụng HPLC đầu dò UV hay PDA, GC, GC, LC-MS, GC-MS nên được thực hiện trong việc xác định tạp bằng qui trình phân tích đã được thẩm định [5].
Theo như các yêu cầu hiện hành và những hạn chế gần đây, nghiên cứu lão hóa trong điều kiện khắc nghiệt, nghiên cứu độ tinh khiết pic và cân bằng khối cho phương pháp định lượng tạp chất nên được thực hiện. Cân bằng khối là “quá trình cùng thêm giá trị định lượng và mức sản phẩm phân hủy để xem mức độ tiệm cận của tổng này so với 100% giá trị ban đầu, với sự xem xét kỹ lưỡng biên độ của độ chính xác phân tích” [15].
Các tạp mà được xác định trên ngưỡng định danh nên được xem xét trong việc đánh giá hồ sơ tạp chất cho dược chất/sản phẩm thuốc.
1.2.2. Tiêu chuẩn chất lượng dược chất [2]
Liều dùng hằng ngày tối đa: ≤ 2 g/ngày: 0.15% hoặc 1.0 mg trên ngày (lấy giá trị nào thấp hơn)
Liều dùng hằng ngày tối đa: > 2 g/ngày: 0.05%.
Việc thực hiện các nghiên cứu tương thích hoạt chất với mỗi tá dược là một trong các cách tiếp cận được khuyến nghị để đánh giá các con đường phân hủy trong sản phẩm thuốc. Các nghiên cứu này nên được thiết kế theo như tài liệu sẵn có và những tương tác đã được biết đến cho vài tá dược như Povidon, Silicon dioxid, lactose, polysorbat, etc [14] [15].
Các điều kiện oxy hóa và ánh sáng yêu cầu việc cân nhắc cụ thể để hiểu về cơ chế của sự phân hủy ánh sáng và khả năng cho độc tính từ tạp phân hủy trong điều kiện ánh sáng [18]. Thử nghiệm độ ổn định điều kiện ánh sáng nên được thực hiện theo như ICH Q1B như một phần không thể thiếu của thử nghiệm lão hóa điều kiện khắc nghiệt [13].
1.2.3. Tiêu chuẩn chất lượng sản phẩm thuốc [3]
Liều dùng tối đa hằng ngày: <10 mg: 1.0% hay 50 µg của tổng liều dùng hằng ngày (lấy giá trị thấp hơn).
Liều dùng tối đa hằng ngày: 10 mg – 100 mg: 0.5% hay 200 µg của tổng liều dùng hằng ngày (lấy giá trị thấp hơn).
Liều dùng tối đa hằng ngày: > 100 mg – 2 g: 0.2% hay 3 mg của tổng liều dùng hằng ngày (lấy giá trị thấp hơn).
1.3. Tạp đồng phân
Đồng phân đối quang là đồng phân lập thể mà hình ảnh phản chiếu của nó qua gương không chồng lên nhau có các tính chất lý-hóa giống hệt nhau ngoại trừ chúng quay mặt phẳng của ánh sáng phân cực theo các hướng ngược nhau và với lượng bằng nhau [16].
Nếu một trong các chất đối quang được coi là hoạt chất, thì chất kia có thể/sẽ được coi là tạp chất/ chất không mong muốn.
Trước đây, người ta không tập trung nhiều vào các đồng phân đối quang riêng lẻ và chưa được nghiên cứu hoặc xác định đặc tính cụ thể do hạn chế của kỹ thuật phân tích. Các kỹ thuật tiên tiến hiện nay cho phép xác định, nghiên cứu hoàn chỉnh, phân tách, và định lượng các tạp chất đồng phân lập thể [17].
Nếu có thể, cần nghiên cứu độ tinh khiết đồng phân lập thể của nguyên liệu ban đầu bằng quy trình phân tích có tính chọn lọc lập thể có độ phân giải cao và đã được thẩm định, đồng thời cũng nên thiết lập khả năng hình thành của tâm lập thể trong các mô tả chi tiết và các kiểm soát của mỗi quá trình tổng hợp [19].
Đối với phương pháp theo dõi độ ổn định của dược chất, cần phải thực hiện các nghiên cứu lão hõa điều kiện khắc nghiệt đối với các phương pháp tạp chất bất đối xứng và xác nhận sự hình thành của nó. Nếu tạp chất bất đối làm tăng các nghiên cứu về tính không ổn định, thì nó cần được theo dõi trong các sản phẩm thuốc.
1.3.1. Kỹ thuật phân tích
Các kỹ thuật phân tích tiên tiến được sử dụng trong phân tách bất đối xứng là Sắc ký khí (GC), Sắc ký lỏng hiệu năng cao (HPLC), UHPLC, Điện di mao quản (CE) và Quang phổ cộng hưởng từ hạt nhân pha lỏng (NMR). Xu hướng xuất hiện gần đây của pha tĩnh bất đối có nguồn gốc Polysaccharic (CSP) đã được công nhận là vật liệu mạnh nhất để tách sắc ký các chất đối quang trong các ứng dụng phân tích và điều chế do lĩnh vực ứng dụng rộng rãi và khả năng chịu tải đáng kể của chúng, nhiều loại dung môi cả phân cực và không phân cực có thể được sử dụng để đạt được sự phân tách mong muốn.
1.3.2. Tiêu chuẩn chất lượng sản phẩm thuốc
Liều dùng tối đa hằng ngày: ≤ 2 g/ngày: 0.15% hay 1.0 mg theo ngày (lấy giá trị thấp hơn).
Liều dùng tối đa hằng ngày: > 2 g/ngày: 0.05%.
Trong hầu hết các trường hợp, tạp chất không đối xứng được kiểm soát trong dược chất nhưng trong một số trường hợp, việc kiểm soát tạp chất không đối xứng và xác nhận sự thay đổi không thể chấp nhận được về độ tinh khiết lập thể hoặc tỷ lệ của hoạt chất xảy ra trong thời hạn sử dụng thuốc phải được thiết lập bằng phương pháp phân tích đã được thẩm định [20].
1.3.3. Tiêu chuẩn chất lượng sản phẩm thuốc
Nói chung, không bao gồm trong tiêu chuẩn chất lượng, nhưng trong một số trường hợp, đánh giá có thể được yêu cầu.
2. Tạp chất vô cơ
Các tạp chất vô cơ có thể là kết quả của quá trình sản xuất. Chúng thường đã được biết đến và được xác định, trong đó bao gồm thuốc thử, phối tử và chất xúc tác, kim loại nặng hoặc các nguyên liệu tồn dư khác, muối vô cơ, các vật liệu khác (ví dụ: chất trợ lọc, than hoạt tính) [1]. Kiểm tra kim loại nặng bằng cách so màu là một phần của USP kể từ năm 1904, do nhiều vấn đề về sức khỏe của bệnh nhân đã xảy ra, việc kiểm soát các tạp chất kim loại có hiệu lực và được kiểm soát hoàn toàn từ ngày 1 tháng 1 năm 2018 trên tất cả các cơ quan quản lý.
2.1. Tạp chất nguyên tố
Dựa trên nhiều thập kỷ nghiên cứu, cứng với kiến thức về tác dụng độc tính và để cải thiện tính an toàn và hiệu quả của thuốc, các yêu cầu hiện nay đã thay đổi và các quy trình phân tích kim loại nặng không đặc hiệu được thay thế bằng các kỹ thuật định lượng đặc hiệu [21] và các tiêu chuẩn cũng được đưa ra [22]. Thử nghiệm so sánh màu cổ điển trong dược điển thiếu khả năng phát hiện một vài tạp chất kim loại. Các dung dịch chuẩn của tất cả các kim loại cũng có khả năng hòa tan khác nhau do mức độ phản ứng của kim loại với ion sunfua có thể thay đổi, điều này cũng tạo ra một vấn đề so sánh trực quan với dung dịch chuẩn chì như trong Hình 5 [23].

Chất xúc tác quá trình và chất gây nhiễm từ môi trường có thể xuất hiện trong dược chất, tá dược hoặc sản phẩm thuốc. Các tạp chất này có thể xuất hiện một cách tự nhiên, hay được thêm vào một cách có chủ ý hoặc vô tình. Vì các tạp chất nguyên tố không mang lại bất kỳ lợi ích điều trị nào cho bệnh nhân, ngoài việc tiếp xúc liên tục có thể gây độc (vì độc tính liên quan đến việc kéo dài thời gian tiếp xúc), mức độ độc hại của chúng trong sản phẩm thuốc cần được kiểm soát trong giới hạn có thể chấp nhận được [24].
3.1.1. Đánh giá rủi ro
Tạp chất nguyên tố được phân loại thành 4 loại [1, 2A, 2B, và 3] dựa trên đường dùng, các giới hạn và yêu cầu kiểm soát khác nhau. Đánh giá rủi ro nên được thực hiện đối với các nguyên tố: Cd, Pb, As, Hg, Co, V, Ni, Tl, Au, Pf, Ir, Os, Ph, Ru, Se, Ag, Pt, Li, Sb, Ba, Mo, Cu, Sn, Cr theo ICH Q3D [6] và USP <232> dựa trên nguồn tiềm ẩn, đánh giá dữ liệu độc tính liên quan đến đường sử dụng, thiết lập mức phơi nhiễm hàng ngày cho phép (PDE) và các giải thích đưa ra cho các mức cao hơn của PDE đã được thiết lập.
Trong đánh giá rủi ro sản phẩm thuốc, các nguồn tạp chất nguyên tố tiềm ẩn cần được xem xét là các nguyên tố được thêm vào một cách có chủ ý như chất xúc tác quá trình, các nguyên tố có thể có trong nguyên liệu được sử dụng để pha chế sản phẩm thuốc và các nguyên tố có thể được đưa vào từ thiết bị sản xuất hoặc hệ thống bao bì [24] [6].
Dựa trên đánh giá rủi ro, cần có biện pháp kiểm tra các tạp chất nguyên tố trong thử nghiệm thường quy đối với các thành phần của thuốc hoặc đối với thành phẩm thuốc, nói cách khác nếu các tạp chất nguyên tố được kiểm soát trong nguyên liệu ban đầu (hoạt chất hay tá dược) như một phần của thử nghiệm thường quy, thì việc kiểm tra thành phẩm thường quy có thể không cần thực hiện. ICH Q3D xác định các phương án khác nhau (Phương án 1, Phương án 2a, Phương án 2b và Phương án 3) để đánh giá rủi ro và chiến lược kiểm soát (nếu kết quả phân tích theo lô luôn ít hơn 30% PDE thì không cần kiểm soát bổ sung).
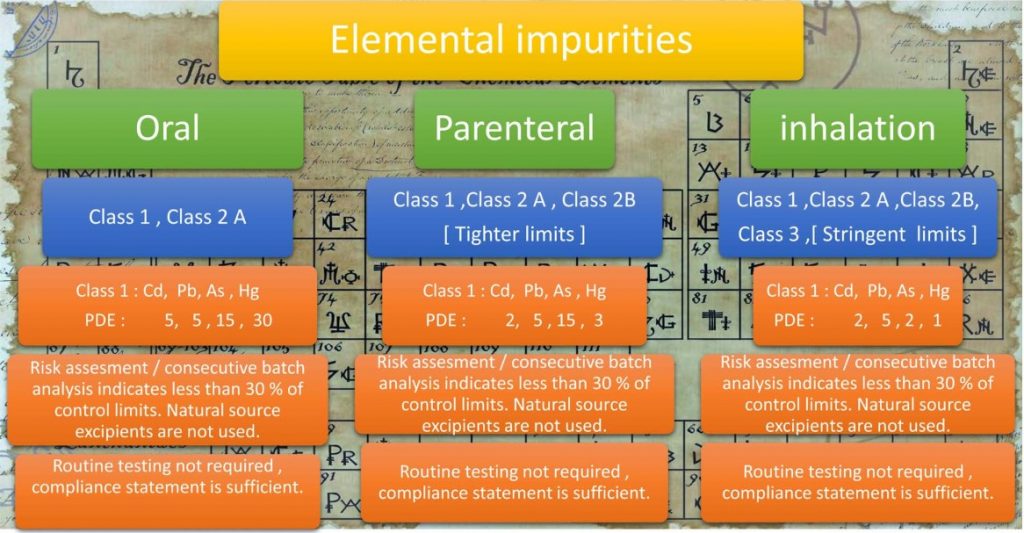
Tổng quan về các tạp chất nguyên tố theo ICH Q3D cần xem xét để đánh giá rủi ro, được trình bày Hình 6 bên dưới.
3.1.2. Tiêu chuẩn chất lượng
ICH Q3D phân loại tạp chất kim loại thành Loại 1, 2A, 2B và 3 với mức độ độc hại giảm dần. Các tiêu chuẩn chất lượng và loại tạp chất cần kiểm soát khác nhau theo đường sử dụng.
3.1.3. Kỹ thuật phân tích
Nói chung, các kỹ thuật phân tích điển hình được áp dụng để phân tích tạp chất nguyên tố là ICP, ICP-MS. Chúng có thể được phân tích bằng phương pháp được liệt kê trong USP <233>, nếu các phương pháp thay thế được sử dụng thì chúng phải được xác thực theo ICH Q2, USP <1225>. Có rất ít phương pháp phân tích thay thế có sẵn và được báo cáo trong các bài báo nghiên cứu [23].
4. Dung môi tồn dư
Dung môi tồn dư là chất lỏng hữu cơ được sử dụng làm phương tiện để chuẩn bị dung dịch hoặc hỗn dịch trong quá trình tổng hợp dược chất hoặc tá dược hoặc trong việc pha chế thuốc.
Dung môi tồn dư được sử dụng trong mỗi giai đoạn của quá trình sản xuất dược chất hoặc tá dược có thể được phân tích và chứng tỏ rằng chúng đáp ứng các yêu cầu của ICH Q3C [7], USP <467> [8] hoặc chúng có thể được phân tích trong giai đoạn cuối cùng, trong cả hai trường hợp, chúng phải được phân tích bằng phương pháp đã xác thực hoặc phương pháp được chỉ định trong USP <467> [8]. Các dung môi tồn dư với các giới hạn đã biết và chi tiết về độc tính liên quan đến loại của chúng đã được liệt kê trong ICH Q3C, các thông số kỹ thuật của chúng trong sản phẩm cuối cùng phải được duy trì ở mức thấp nhất có thể (Các giới hạn chấp nhận phải dựa trên ICH Q3C, USP <467>). Nếu sử dụng các dung môi không được liệt kê, chúng phải được kiểm soát theo giới hạn độc tính của dung môi tương ứng.
Trong bản sửa đổi gần đây của ICH Q3C, Triethylamine được thêm vào làm dung môi Loại 3 với giới hạn PDE (62,5 mg/ ngày) và giới hạn cho Methyl isobutyl ceton được sửa đổi (Loại 2) với giới hạn PDE (45 mg/ ngày) theo dữ liệu độc tính mới.
4.1. Kỹ thuật phân tích
Thông thường, các phương pháp GC và GC-HS được sử dụng và trong một số trường hợp, HPLC, IC và các kỹ thuật khác được sử dụng. Nếu phương pháp dung môi tồn dư được chỉ định trong chuyên luận dược điển thì có thể tuân theo phương pháp tương tự hoặc có thể sử dụng phương pháp được chỉ định trong USP <467>. USP <467> không chỉ định phương pháp cho dung môi loại 3 nhưng FDA trích dẫn để kiểm soát và báo cáo dung môi loại 3 cũng bằng một phương pháp đã được thẩm định.
4.2. Tiêu chuẩn chất lượng dược chất
Theo phân loại ICH Q3C.
4.3. Tiêu chuẩn chất lượng sản phẩm thuốc
Vì giới hạn dung môi tồn dư đã được thiết lập theo hướng dẫn và được định lượng trong tá dược và dược chất riêng lẻ, nên không cần kiểm tra dung môi tồn dư trong thành phẩm thuốc. Sự tuân thủ có thể được chứng minh thông qua đánh giá rủi ro một cách toàn diện dựa trên kết quả định lượng của dung môi tồn dư có trong các dược chất và tá dược riêng lẻ [25].
Nếu dung môi tồn dư được sử dụng trong quá trình sản xuất các sản phẩm thuốc, chúng phải được định lượng bằng các phương pháp phân tích đã được thẩm định với giới hạn của ICH Q3C.
Nếu sử dụng dung-môi-không-được-liệt-kê trong quy trình, thì nên trình bày/ thực hiện các biện luận cho các giới hạn được đề xuất, các chiến lược kiểm soát và đánh giá độc tính.
5. Dạng/tạp đa hình
Đánh giá tính đa hình sẽ được sử dụng để định tính và định lượng các chất vô định hình và tinh thể theo USP <941>, ICH Q6A, và hướng dẫn của FDA “ANDAs: Tính đa hình dược chất dạng rắn” [26] [27].
Do các đặc tính vật lý của nguyên liệu ban đầu có thể thay đổi theo dạng đa hình, ảnh hưởng đến quá trình sản xuất dược chất và sản phẩm thuốc, cũng như tính ổn định, độ hòa tan và sinh khả dụng của sản phẩm, loại và mức độ của các đặc tính và thử nghiệm giải phóng được thực hiện trên đồng tinh thể phải đủ để đảm bảo nhận dạng, độ bền, chất lượng và độ tinh khiết của (các) API và độ ổn định của sản phẩm thuốc [54] [55] [56]. Bất kỳ dạng đa hình nào khác với dạng đa hình mong muốn sẽ được coi là tạp chất [57].
Dựa trên quy trình và đánh giá được thực hiện bởi nhà sản xuất dược chất dạng đa hình sẽ được xác nhận.
Trong sản phẩm thuốc, dạng đa hình dược chất không được thay đổi do quy trình sản xuất sản phẩm thuốc hoặc do tá dược hoặc do bảo quản trong thời gian dài. Các mẫu ban đầu của sản phẩm thuốc và các mẫu độ ổn định phải được đánh giá để chứng minh tính ổn định của dạng đa hình trong suốt thời hạn sử dụng [27]. Thông tin về các dạng đa hình khác nhau của các dược chất thường được đề cập trong các bằng sáng chế tương ứng.
5.1. Kỹ thuật phân tích
Kỹ thuật phân tích phổ biến được sử dụng là phương pháp nhiễu xạ tia X. Phương pháp được áp dụng phải cụ thể đối với dạng tinh thể mong muốn của hợp chất dựa trên quy trình sản xuất dược chất. Phương pháp phải được xác thực theo USP <1225> [5].
Trong một số trường hợp, mẫu XRD của các dạng đa hình khác nhau có thể giống nhau, khi đó các kỹ thuật khác như Phân tích nhiệt (DSC) hoặc quang phổ Raman nên được sử dụng.
6. Tạp chất gây độc gen (có khả năng gây đột biến và gây ung thư)
Tạp chất gây độc gen là các chất phản ứng DNA có khả năng trực tiếp gây ra tổn thương DNA khi hiện diện ở mức thấp dẫn đến đột biến và do đó, có khả năng gây ung thư. Loại chất gây ung thư đột biến này thường được phát hiện trong một xét nghiệm gây đột biến ngược (gây đột biến) của vi khuẩn. Đánh giá dựa trên cấu trúc rất hữu ích để dự đoán kết quả gây đột biến gen của vi khuẩn dựa trên kiến thức đã được thiết lập. Có nhiều cách tiếp cận khác nhau để tiến hành đánh giá này, bao gồm xem xét các tài liệu hiện có và / hoặc đánh giá độc chất bằng mô hình toán học [60].
Định nghĩa của ICH M7 về tạp chất gây độc gen không áp dụng cho các dược chất và sản phẩm thuốc dành cho các chỉ định ung thư giai đoạn muộn như được định nghĩa trong phạm vi của ICH S9 [10] [11]. Ngoài ra, có thể có một số trường hợp dược chất dùng cho các chỉ định khác tự gây độc cho gen ở nồng độ điều trị và có thể được cho là có liên quan đến việc tăng nguy cơ ung thư. Việc tiếp xúc với tạp chất gây đột biến trong những trường hợp này sẽ không làm tăng đáng kể nguy cơ ung thư của dược chất. Do đó, các tạp chất có thể được kiểm soát ở mức chấp nhận được đối với các tạp chất không gây đột biến [29].
Các tạp chất dưới giới hạn định danh với các cảnh báo về cấu trúc (ví dụ, hợp chất N-nitroso-, và alkyl-azoxy), và các tạp chất tiềm ẩn nên được xem xét để đánh giá gây độc gen/ gây đột biến [28]. Việc đánh giá dựa trên rủi ro này đối với các quá trình tổng hợp riêng lẻ nên được thực hiện bằng cách xem xét nguyên liệu ban đầu / thuốc thử được sử dụng trong quá trình và các sản phẩm trung gian/ sản phẩm phụ được hình thành trong quá trình tổng hợp [29]. Sự phù hợp của chiến lược kiểm soát được đề xuất có thể được hỗ trợ bằng thông tin về bất kỳ tạp chất gây đột biến nào được hình thành hoặc loại bỏ trong các bước sản xuất giữa nguyên liệu ban đầu được đề xuất và dược chất, hoặc được kiểm soát trong đặc điểm kỹ thuật của nguyên liệu ban đầu được đề xuất [28].
6.1. Kỹ thuật phân tích
LCMS, GC-MS thường được sử dụng để phát hiện và định lượng tạp chất độc gen nhằm đạt được giới hạn định lượng mong muốn. Các phương pháp HPLC, UPLC và GC có thể được sử dụng trong một số trường hợp.
6.2. Ngưỡng quan tâm độc tính (TTC)
Khái niệm ngưỡng quan tâm độc tính (TTC) được phát triển để xác định một lượng chấp nhận được đối với bất kỳ hóa chất chưa được nghiên cứu nào có nguy cơ gây ung thư hoặc các tác dụng độc hại khác không đáng kể. Các phương pháp mà TTC dựa trên thường được coi là rất thận trọng vì chúng liên quan đến phép ngoại suy tuyến tính đơn giản từ liều lượng cho tỷ lệ mắc khối u 50% (TD50) đến 1 trong 106 trường hợp mắc bệnh, sử dụng dữ liệu TD50 cho các loài nhạy cảm nhất và vị trí nhạy cảm nhất của cảm ứng khối u [10].
Giới hạn nồng độ tính bằng ppm của tạp chất gây độc gen trong dược chất bắt nguồn từ ngưỡng quan tâm về độc tính (TTC) có thể được tính toán dựa trên liều hàng ngày dự kiến cho bệnh nhân bằng công thức [10] [30].
Giới hạn nồng độ (ppm) = TTC [μg/ngày]/liều (g/ngày)
Tổng quan về hướng dẫn tạp chất gây độc gen được trình bày trong Hình 7.
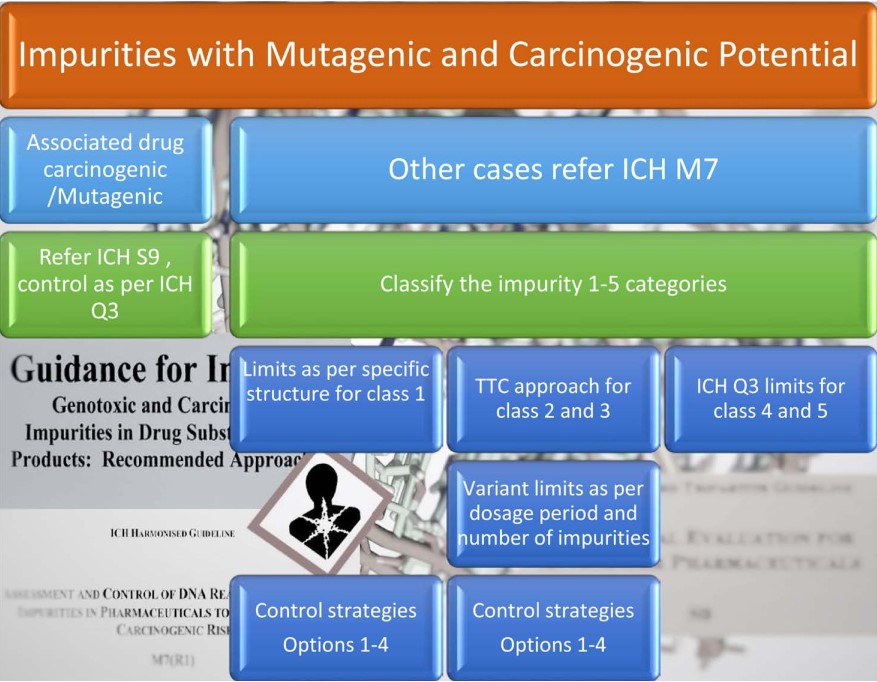
Hướng dẫn ICH M7 phân loại các tạp chất có thể gây độc gen thành 5 loại, được trình bày trong Bảng 1 dưới đây.
Đánh giá rủi ro tiêu chuẩn của các chất gây ung thư đã biết giả định rằng nguy cơ ung thư tăng lên như một hàm của liều tích lũy. Do đó, nguy cơ ung thư của liều thấp liên tục trong suốt cuộc đời sẽ tương đương với nguy cơ ung thư liên quan đến mức phơi nhiễm tích lũy giống hệt nhau được tính trung bình trong một thời gian ngắn hơn. ICH M7 xác định các giới hạn cho một tạp chất đơn lẻ và cho tổng số hai tạp chất trở lên theo thời gian điều trị theo các giới hạn dưới đây của Hình 8 [10].
Bảng 1. Tạp chất độc tính lên gen theo ICH M7 được phân thành 5 loại [10]

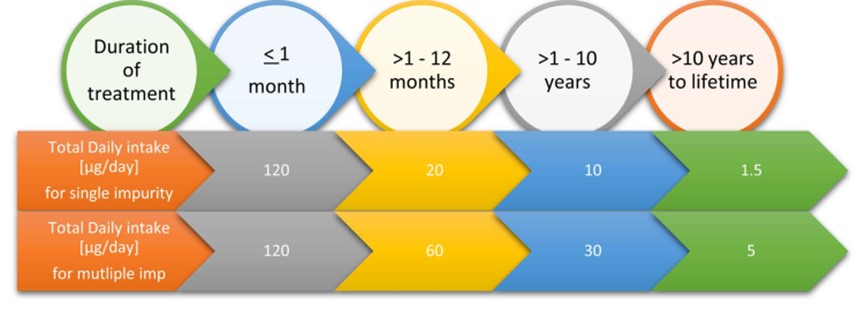
ICH M7 xác định các chiến lược điều khiển khác nhau với Tùy chọn 1-5. Phương án 3 cho phép các nhà sản xuất tránh việc kiểm tra xuất xưởng nếu tạp chất ít hơn 30% mức kiểm soát trong một vài lô.
Các hãng dược đang chấp nhận khái niệm này một cách từ từ tuy nhiên các cơ quan quản lý đang theo dõi và công bố các thiếu sót để kiểm soát các tạp chất. Tạp chất bromochloropropionphenon trong Bupropion Hydrochloride, USP chỉ định tạp chất này ở không quá 0,1% nhưng theo tài liệu tạp chất phải được kiểm soát ở 4 ppm, FDA đã trích dẫn đây là sự thiếu hụt đối với việc biên tập.
6.3. Tiêu chuẩn chất lượng
Theo ICH M7 và S9 hoặc các giới hạn độc tính tạp chất tương ứng.
7. Từ khóa và nguồn
Từ khóa
Hồ sơ tạp chất, sự phân hủy, đồng phần đối quang, tính độc lên gen, đa thù hình, tạp nguyên tố, tạp hữu cơ, tạp vô cơ, tạp chất, tiêu chuẩn chất lượng.
Nguồn
Nội dung chính của bài đăng này được tham khảo và trích dẫn từ tác giả “Pannala, R. (2018), Impurity Profiling of Solid Oral Drug Products to Sail through GDUFA-II Requirements, American Journal of Analytical Chemistry, 9, 187-209”.
8. Tài liệu tham khảo
- USP , Impurities in Drug substances and drug products.
- ICH Q3A Impurities in New Drug Substances.
- ICH Q3B Impurities in New Drug Products.
- ICH Q1A Stability Testing of New Drug Substances and Products.
- ICH Q2 Validation of Analytical Procedures.
- ICH (2014) Guideline for Elemental Impurities Q3D. ICH Harmonized Guideline.
- ICH Q3C Impurities: Residual Solvents
- USP Residual Solvents.
- ICH Q6A Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances
- ICH M7 Assessment and Control of DNA Reactive (Mutagenic) Impurities in pharmaceuticals to Limit Potential Carcinogenic Risk.
- ICH S9 Nonclinical Evaluation for Anticancer Pharmaceuticals.
- Sharp, T.R. (Presentation) Forced Degradation: What? Why? How? FreeThink Technologies, Inc., Groton, Connecticut.
http://www.cbinet.com/sites/default/files/files/Workshop%20B(3).pdf - ICH Q1B, Photostability Testing of New Substances and Products
- Fathima, N., Mamatha, T., Qureshi, H.K., Anitha, N. and Rao, J.V. (2011), Drug-Excipient Interaction and Its Importance in Dosage form Development. Journal of Applied Pharmaceutical Science, 1, 66-71.
- Bharate, S.S., Bharate, S.B. and Bajaj, A.N. (2010) Interactions and Incompatibilities of Pharmaceutical Excipients with Active Pharmaceutical Ingredients: A Comprehensive Review. Journal of Excipients and Food Chemicals, 1, 3-26.
- European Medicines Agency (1994) Investigation of Chiral Active Substance. European Medicines Agency Guideline.
- FDA (1992) Development of New Stereoisomeric Drugs. FDA Guidance.
- Janzen, H. (2016) Forced Degradation Studies—Comparison between ICH, EMA, FDA and WHO Guidelines and ANVISA’s Resolution RDC 53/2015. Master of Drug Regulatory Affairs, Rheinische Friedrich-Wilhelms-University of Bonn, Bonn.
- Karnes, H.T. and Sarkar, M.A. (1987) Enantiomeric Resolution of Drug Compounds by Liquid Chromatography. Pharmaceutical Research, 4, 285- 292.
https://doi.org/10.1023/A:1016437018323 - Canada Health (2000) Guidance on Stereochemical Issues in Chiral Drug Development.
- USP Elemental Impurities-Procedures.
- USP Elemental Impurities-Limits.
- Raghuram, P., Soma Raju, I.V. and Sriramulu, J. (2010) Heavy Metals Testing in Active Pharmaceutical Ingredients: An Alternate Approach. Pharmazie, 65, 15-18.
- CDER (2016) Elemental Impurities in Drug Products Guidance for Industry. Draft Guidance
- CDER (2009) Guidance for Industry Residual Solvents in Drug Products Marketed in the United States
- USP X-Ray Powder Diffraction.
- CDER (2007) Guideline on ANDAs: Pharmaceutical Solid Polymorphism Chemistry, Manufacturing, and Controls Information.
- Benigni, R. and Bossa, C. (2006) Structural Alerts of Mutagens and Carcinogens. Current Computer-Aided Drug Design, 2, 169-176.
http://www.iss.it/binary/meca/cont/Ccadd2006%20.1161263198.pdf - ICH Q11 Guideline: Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities). Questions and Answers Version, 23 August 2017.
- EMEA (2007) Guideline on the Limits of Genotoxic Impurities.
- Pannala, R. (2018) Impurity Profiling of Solid Oral Drug Products to Sail through GDUFA-II Requirements. American Journal of Analytical Chemistry, 9, 187-209.



You must be logged in to post a comment.